
Woman With Weakness, Diplopia and Dysphagia
What's Your Diagnosis?
Sharpen Your Physical Diagnostic Skills
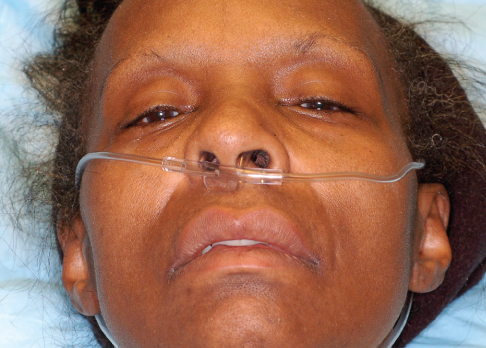
HISTORY
A 58-year-old woman complains of generalized weakness, diplopia, and dysphagia. Symptoms began 1 week earlier and have progressively worsened. She can no longer walk without assistance. Has not been able to swallow solid foods for the past 3 days and now has difficulty in swallowing her own saliva. Also has difficulty in opening her eyelids completely. She says she feels as if she is unable to take a deep breath. No fevers, recent illness, or new medications.
Past medical history is significant for hypothyroidism (for which she takes levothyroxine), chronic
obstructive pulmonary disease, and gastric bypass surgery (performed 10 years earlier).
PHYSICAL EXAMINATION
Patient is awake and alert. She is in no acute distress but clearly has difficulty in swallowing and is spitting, rather than swallowing, secretions. She has bilateral ptosis and bilateral facial paresis; tongue musculature is weak with no fasciculations. Extraocular muscles are abnormal with impairment of left eye abduction and elevation. Sensation to light touch, vibration, and temperature are normal and equal throughout. Lower extremity strength is 2/5 bilaterally, upper extremity strength is 2/5 proximally and 3/5 distally. Lower extremity reflexes are absent; upper extremity reflexes decreased. Gait could not be assessed because of weakness.
WHAT'S YOUR DIAGNOSIS?
What's Your Diagnosis?
ANSWER: MYASTHENIA GRAVIS
The patient has myasthenia gravis, an autoimmune neuromuscular disease characterized by skeletal muscle weakness and fatigability. IgG autoantibodies interact with the postsynaptic acetylcholine receptors (AChR) at the nicotinic neuromuscular junction, reducing the number of normally functioning AChRs. The disease is thought to result from either dysfunction of the thymus or immune response to exogenous antigens. Abnormalities of the thymus gland (typically thymoma or thymic hyperplasia) have been found in 75% of patients with myasthenia gravis.
The disease affects 1 in 10,000 persons and is generally seen in younger women (20 to 30 years old) and older men (60 to 70 years old).
Patients often present with generalized weakness of the limbs and the facial, bulbar, and ocular muscles. Fewer than one-fifth of patients have symptoms confined to the eyes (diplopia and ptosis). In more severe cases, the respiratory muscles can be affected and the patient may require mechanical ventilation.
DIAGNOSIS
Myasthenia gravis can be diagnosed using a combination of pharmacologic testing, electromyography (EMG), and a laboratory test for the detection of anti-AChR antibodies. The edrophonium (Tensilon) test is the standard pharmacologic test used. Edrophonium is an acetylcholinesterase inhibitor which, when administered to a patient with a myasthenic crisis, should increase muscle strength 30 seconds after administration. However, a negative test cannot be used to exclude myasthenia gravis.
In patients with myasthenia gravis, EMG will demonstrate a characteristic rapid reduction in the muscle action potential with repetitive nerve stimulation. AChR antibody testing is the most specific test, but patients with mild disease may have undetectable titers.
Imaging of the mediastinum is often done to assess the thymus gland. If there is concern about respiratory muscle involvement, pulmonary function testing (such as a negative inspiratory force [NIF] measurement) should be done.
TREATMENT
Treatment of myasthenia gravis includes acetylcholinesterase inhibitors, chronic immune suppression with corticosteroids or azathioprine, and possibly thymectomy. Acetylcholinesterase inhibitors such as pyridostigmine and neostigmine help improve neuromuscular transmission and increase muscle strength. Immunosuppressants can improve muscle strength by suppressing the production of the AChR antibodies. Thymectomy is an effective treatment for patients with a thymoma, and it can increase the probability of remission in non-thymomatous autoimmune myasthenia gravis.
For a more severe, acute crisis, intravenous immunoglobulin or plasmapheresis (to reduce the amount of circulating antibodies) plus high-dose corticosteroids can be used.
OUTCOME OF THIS CASE
This patient was directly admitted to the hospital and was subsequently intubated because of respiratory failure. She was treated with prednisolone, intravenous immunoglobulin, and pyridostigmine. Her symptoms
improved with treatment; she was extubated and ultimately discharged to a skilled nursing facility, where she received physical and occupational therapy.
DIFFERENTIAL DIAGNOSIS
Guillain-Barré syndrome (GBS) is an acute inflammatory demyelinating polyneuropathy that is characterized by progressive symmetric ascending muscle weakness, paralysis, and hyporeflexia. There may be sensory or autonomic symptoms, and the cranial nerves can be affected as well. GBS is believed to result from the autoimmune responses to a recent infection. Lumbar puncture with spinal fluid analysis can help make the diagnosis. An elevated cerebrospinal fluid protein level with a normal white blood cell count is often seen. The treatment is plasma exchange therapy or intravenous immune serum globulin.
Botulism is caused by the neurotoxin of Clostridium botulinum. It is characterized by a progressive,
symmetric, descending paralysis with initial symptoms that include diplopia, dysphagia, and dysarthria. On physical examination, the pupils are usually dilated and non-reactive, which distinguishes it from myasthenia gravis (in which the pupils are not affected). The diagnosis is made by excluding other causes. Although serum and stool tests for the toxin exist, they are not widely available. Treatment consists of respiratory support if needed and botulism immune globulin.
Lambert-Eaton myasthenic syndrome is an autoimmune disorder that causes muscle weakness and fatigue. In contrast to myasthenia gravis, sustained exercise can increase muscle strength in patients with Lambert-Eaton syndrome. The syndrome is strongly associated with malignancy (particularly small cell lung cancer), and it rarely progresses to respiratory and bulbar failure. EMG is used to confirm the diagnosis. Treatment is supportive, and immunosuppressants can be used to reduce symptom severity. ■
FOR MORE INFORMATION:
■ El-Bawab H, Hajjar W, Rafay M, Bamousa A, Khali A, Al-Kattan K. Plasmapheresis before thymectomy in myasthenia gravis: routine versus selective protocols. Eur J Cardiothorac Surg. 2009;35:392-397.
■ Gronseth GS, Barohn RJ. Thymectomy for myasthenia gravis. Curr Treat Options Neurol. 2002;4(3):203-209.
■ Sloan, E, Handel D, Gaines S. Chronic neurologic disorders. In: Tintinallli J, Stapczynski J, Ma OJ, et al. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York: McGraw Hill; 2011:1166-1170.
■ Thavasothy M, Hirsch N. Myasthenia gravis. Br J Anaesth CEPD Rev. 2002;2(3):88-90.