
Peer Reviewed
A Collection of Conditions Affecting the Respiratory System
Proteus Syndrome With Pulmonary Involvement
Authors:
Min Qiao, MD; Bisrat Haile, MD; Waleed Ali, MD; Kiran Reddy, MD; and Rajat Mukherji, MD
Kingsbrook Jewish Medical Center, Brooklyn, New York
Citation:
Qiao M, Haile B, Ali W, Reddy K, Mukherji R. Proteus syndrome with pulmonary involvement. Consultant. 2016;56(12):1114-1115.
A 23-year-old man was admitted to the hospital with a fever, productive cough, and acute respiratory distress.
Upon admission, a chest radiograph revealed hyperlucency of the right hemithorax with a right basilar infiltrate (Figure 1). Computed tomography (CT) of the chest showed extensive unilateral bullous emphysema involving the entire right lung from apex to base (Figure 2). The emphysema was confined to one side and was so severe and voluminous that the mediastinum was pushed far away from its midline alignment with the sternum into the left hemithorax, resulting in compression and atelectasis of the left lung. There was also a right-sided pneumonia superimposed on the background of emphysema.
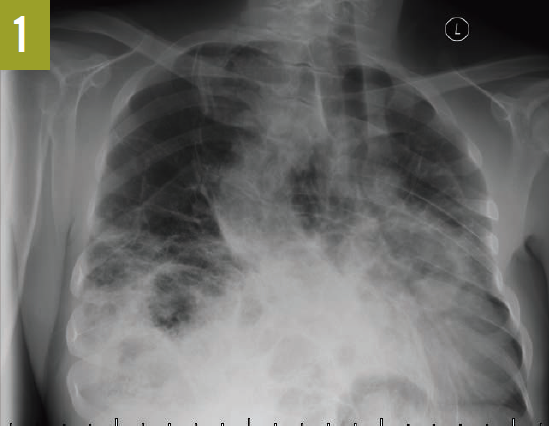
Figure 1. A chest radiograph showed hyperlucency of the right hemithorax and a right basilar infiltrate.
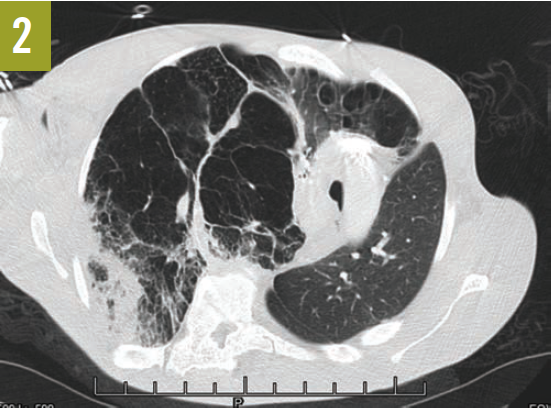
Figure 2. A CT scan of the chest at the level of the carina showed severe and voluminous bullous emphysema of the right lung; the mediastinum is pushed far away from its midline alignment with the sternum into the left hemithorax, and a right basal pneumonia is present.
The patient had an established diagnosis of Proteus syndrome. On physical examination, he exhibited a cerebriform soft tissue nevus on the right palm (Figure 3), which is the result of a proliferation of collagenous tissue in the dermis and is highly specific for Proteus syndrome. Also noted was unilateral hypertrophy of his right hand and foot. A CT scan of the abdomen revealed massive splenomegaly, extending from the diaphragm to the pelvis (Figure 4).
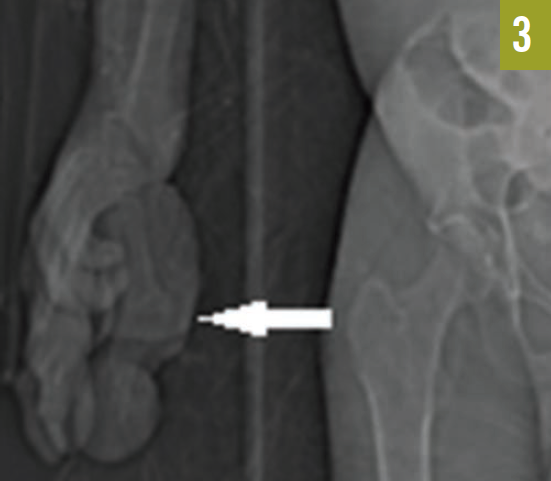
Figure 3. A cerebriform soft tissue nevus was present on the right palm.
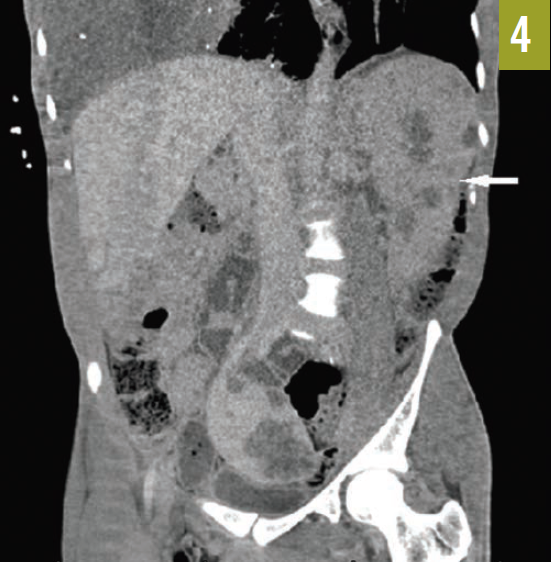
Figure 4. A CT scan of the patient’s abdomen showed massive splenomegaly (arrow).
He received noninvasive mechanical ventilation along with bronchodilators, systemic corticosteroids, and antibiotics. The patient improved quickly with treatment and was subsequently discharged.
Discussion. Proteus syndrome is a rare condition, occurring in less than 1 in 1,000,000 live births.1 It is an entity of disproportional postnatal overgrowth of any organ system or body tissue and most commonly involves bone and skin. The name Proteus comes from the Greek god who is able to transform himself into any shape. It implies the polymorphism of the physical manifestations of the disease. The condition is sporadic and not heritable.2
Persons with the condition are phenotypically normal at birth. The overgrowth of body tissue starts during infancy and progresses rapidly and relentlessly in early childhood, resulting in disruption of normal anatomy and function. The mosaic activating mutation of gene AKT1 49G>A, Glu17Lys, in postzygotic somatic cells was discovered to be the cause of the overgrowth of affected tissue.3,4
Lung involvement affects approximately 10% of patients with Proteus syndrome.5 The clinical manifestations can be either pulmonary venous dilatation or disruptive pulmonary parenchymal overgrowth with subsequent diffuse bullous emphysema.6,7 Pulmonary embolism can be another severe complication, resulting from thrombophilia, the etiology of which is not clear. Patients with lung involvement can have recurrent pneumonia and respiratory failure, with episodes occurring even in early childhood. Treatment is usually directed toward the acute complications of the disease. However, recent research targeting the AKT1 mutation shows promising results.8
Our patient demonstrated an extremely severe form of bullous emphysema confined to the entire right lung. His chest radiograph showed a remarkable unilateral hemithorax hyperlucency, which are findings that can also be seen in another condition, Swyer-James-Macleod (SJM) syndrome.
SJM syndrome is a form of postinfectious bronchiolitis obliterans resulting from childhood pneumonia. The condition is characterized by inflammation and destruction of bronchioles and alveolar buds while they are still in development. The affected lung develops abnormally after the infection and is relatively smaller than the contralateral lung. Radiographs frequently reveal unilateral hyperlucency and decreased vascularity on the affected side. A nuclear scan will show decreased vascular perfusion in the involved lung. A CT scan of the chest usually reveals a small but emphysematous lung.9,10
In Proteus syndrome and SJM syndrome, patients may present with similar pulmonary symptoms and with unilateral hyperlucency visible on chest radiographs. In SJM syndrome, the hyperlucent lung is small or normal in size. Proteus syndrome, on the other hand, demonstrates a hyperlucent lung that is usually large. While SJM syndrome is a consequence of underdevelopment of bronchioles and alveoli after infection, Proteus syndrome is the result of excessive overgrowth of lung tissue. A CT scan usually differentiates one from the other.
References:
- Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C(1):38-52.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14(11):1151-1157.
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611-619.
- Wee JS, Mortimer PS, Lindhurst MJ, Chong H, Biesecker LG, Holden CA. A limited form of Proteus syndrome with bilateral plantar cerebriform collagenomas and varicose veins secondary to a mosaic AKT1 mutation. JAMA Dermatol. 2014;150(9):990-993.
- Newman B, Urbach AH, Orenstein D, Dickman PS. Proteus syndrome: emphasis on the pulmonary manifestations. Pediatr Radiol. 1994;24(3):189-193.
- Lim G-Y, Kim O-H, Kim HW, et al. Pulmonary manifestations in Proteus syndrome: pulmonary varicosities and bullous lung disease. Am J Med Genet A. 2011;155A(4):865-869.
- Launois C, Vallerand H, Perotin JM, et al. The Proteus syndrome: a rare cause of pulmonary emphysema [in French]. Rev Mal Respir. 2013;30(9):789-793.
- Lindhurst MJ, Yourick MR, Yu Y, Savage RE, Ferrari D, Biesecker LG. Repression of AKT signaling by ARQ 092 in cells and tissues from patients with Proteus syndrome. Sci Rep. 2015;5:17162.
- Chaucer B, Chevenon M, Toro C, Lemma T, Grageda M. Swyer-James-Macleod syndrome: a rare finding and important differential in the ED setting. Am J Emerg Med. 2016;34(7):1329.e3-1329.e4.
- Sen HS, Taylan M, Abakay O, Sezgi C, Cetincakmak MG. Adult diagnosis of Swyer-James-Macleod syndrome: retrospective analysis of four cases. Respir Care. 2014;59(4):e51-e54.
NEXT: Pulmonary Hamartoma
Pulmonary Hamartoma
Authors:
Samuel E. Cohen, MD, and Jaime Betancourt, MD
VA Greater Los Angeles Healthcare System, Los Angeles, California
Citation:
Cohen SE, Betancourt J. Pulmonary hamartoma. Consultant. 2016;56(12):1116-1117.
A 62-year-old man with a history of nephrolithiasis presented with dyspnea while walking uphill, which had progressively worsened over the last 10 years. He denied dyspnea with exertion on flat ground. He had never smoked cigarettes.
Diagnostic tests. Chest radiographs revealed complete collapse of the left upper lobe and a compensatory hyperexpanded right lung (Figure 1). Chest computed tomography identified an endobronchial mass in the distal aspect of the left mainstem bronchus (Figure 2), which was negative for any fluorodeoxyglucose uptake on positron emission tomography scan.
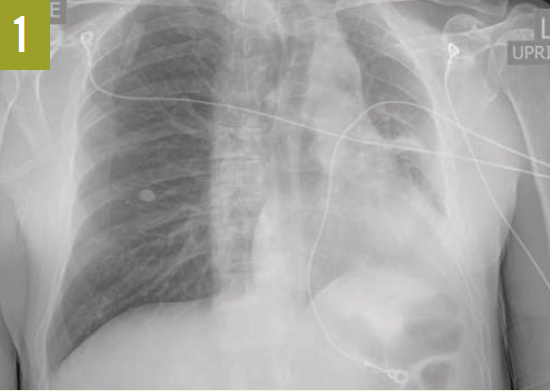
Figure 1. A chest radiograph demonstrated complete collapse of the left upper lobe.
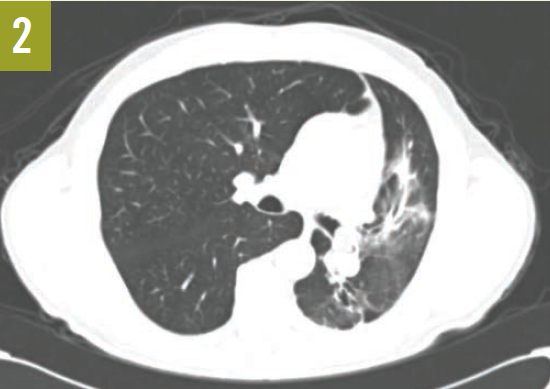
Figure 2. A chest computed tomography demonstrated an endobronchial mass in the distal aspect of the left mainstem bronchus.
Pulmonary function testing demonstrated a moderate obstructive ventilatory defect with evidence of air trapping. Flexible fiberoptic bronchoscopy discovered a soft, fleshy endobronchial mass causing dynamic and near complete obstruction of the left mainstem bronchus, approximately 3.5 cm distal to the carina (Figure 3). Histopathologic evaluation of the tissue biopsy was consistent with a benign hamartoma.
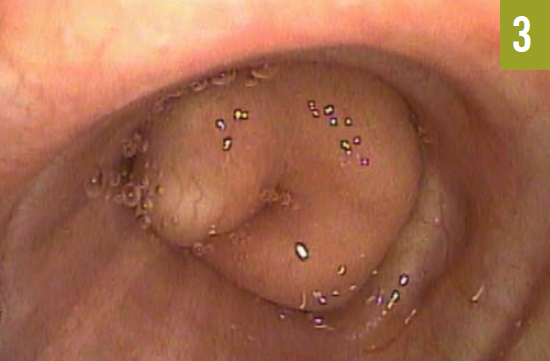
Figure 3. An endobronchial hamartoma was visualized in the left mainstem bronchus with flexible fiberoptic bronchoscopy.
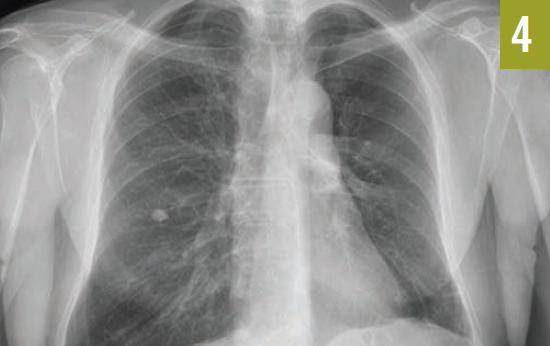
Figure 4. A chest radiograph demonstrated complete reexpansion of the left lung after intervention with laser ablation.
Treatment. The patient underwent rigid bronchoscopy with laser ablation for removal of the endobronchial hamartoma. Postoperative chest radiographs demonstrated complete reexpansion of the left lung (Figure 4).
Discussion. Pulmonary hamartomas are the most common benign tumor of the lung.1-4 They are histologically heterogeneous, often a combination of cartilage, fat, myxomatous tissue, fibroblastic tissue, or muscle.1 They typically grow slowly over many years and are often identified at middle age.1
Pulmonary hamartomas are most commonly located in the parenchyma and are usually of minimal clinical significance.1,2 Rarely, a pulmonary hamartoma can present as an endobronchial mass.1-3,5-7 One study1 found that only 1.4% of pulmonary hamartomas are located within the bronchus. Even when located within the bronchus, many patients remain asymptomatic.3 However, if the lesion grows large enough to obstruct the airway, patients may develop cough, dyspnea, or postobstructive pneumonia.2,3,5 Hemoptysis may also be a presenting symptom.2
The differential diagnosis for endobronchial lesions is broad, and benign and malignant etiologies should be considered. Malignant endobronchial lesions can be categorized as either primary endoluminal carcinoma or metastatic carcinoma to the bronchus. Primary endoluminal carcinoma is typically due to bronchogenic carcinoma, carcinoid tumor, adenoid cystic carcinoma, or mucoepidermoid carcinoma.5
The most common etiologies of metastatic carcinoma to the bronchus are renal cell carcinoma, breast carcinoma, thyroid carcinoma, colon carcinoma, melanoma, and sarcoma.5 Benign endobronchial lesions can be the result of abnormal tissue growth within the lumen, typically in the form of hamartoma, leiomyoma, papilloma, or granulation tissue.5 Foreign bodies, mucus plugs, and blood clots can also result in endobronchial obstruction.5 Symptoms are typically more gradual in onset when the etiology is a tumor compared with an obstruction.5
Treatment of endobronchial hamartomas has historically been with surgical resection; however, technologic advancements have made bronchoscopic intervention a feasible, less-invasive, and safe alternative for removal.7 Surgical resection is now reserved for cases in which endobronchial techniques have been unsuccessful.3 The most common bronchoscopic options for hamartoma removal include neodymium-doped yttrium aluminum garnet (Nd:YAG) laser, electrocautery, and cryotherapy.3,6,7 Traditionally, a rigid bronchoscope is used for intervention, but flexible bronchoscopy has been used increasingly for treatment of endobronchial lesions.3 Our patient was treated with Nd:YAG laser via a rigid bronchoscope.
Outcome of the case. Our patient’s endobronchial hamartoma had grown insidiously over many years, allowing his pulmonary physiology to compensate over time despite worsening airway compromise. This case demonstrates that the clinical importance of a pulmonary hamartoma depends on location. If it is endobronchial, procedural intervention may be required. Since these lesions are benign, outcomes are usually favorable, with a low risk of recurrence.2 This patient’s symptoms resolved, and he is now able to run uphill without limitation.
References:
- Gjevre JA, Myers JL, Prakash UB. Pulmonary hamartomas. Mayo Clin Proc. 1996;71(1):14-20.
- Cosío BG, Villena V, Echave-Sustaeta J, et al. Endobronchial hamartoma. Chest. 2002;122(1):202-205.
- Mondello B, Lentini S, Buda C, et al. Giant endobronchial hamartoma resected by fiberoptic bronchoscopy electrosurgical snaring. J Cardiothorac Surg. 2011;6:97.
- Murray J, Kielkowski D, Leiman G. The prevalence and age distribution of peripheral pulmonary hamartomas in adult males: an autopsy-based study. S Afr Med J. 1991;79(5):247-249.
- Ernst A, Feller-Kopman D, Becker HD, Mehta AC. Central airway obstruction. Am J Respir Crit Care Med. 2004;169(12):1278-1297.
- Jhun BW, Lee K-J, Jeon K, et al. The clinical, radiological, and bronchoscopic findings and outcomes in patients with benign tracheobronchial tumors. Yonsei Med J. 2014;55(1):84-91.
- Sim JK, Choi JH, Oh JY, et al. Two cases of diagnosis and removal of endobronchial hamartoma by cryotherapy via flexible bronchoscopy. Tuberc Respir Dis (Seoul). 2014;76(3):141-145.
NEXT: Pleural Calcifications in a Patient With a History of Tuberculosis
Pleural Calcifications in a Patient With a History of Tuberculosis
Authors:
Vishal V. Patel, MD; Rajat Mukherji, MD; Alok Gupta, MD; Neal Gupta, MD; and Ronak Patel, MBBS
Kingsbrook Jewish Medical Center, Brooklyn, New York
Citation:
Patel VV, Mukherji R, Gupta A, Gupta N, Patel R. Pleural calcifications in a patient with a history of tuberculosis. Consultant. 2016;56(12):1117-1118.
An 87-year-old woman presented to the hospital with recurrent vomiting, which had subsided spontaneously by the time of her arrival. She had a past medical history of atrial fibrillation, coronary artery disease, and diabetes mellitus. In addition, she had a history of pulmonary tuberculosis (TB) in 1939.
Chest radiographs and computed tomography scans taken at admission showed extensive bilateral pleural calcifications with scattered calcified pulmonary nodules throughout both lung fields (Figures 1 and 2).
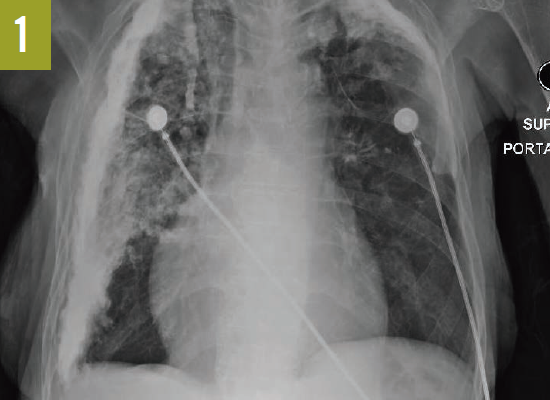
Figure 1. A chest radiograph showed extensive bilateral pleural calcifications with scattered calcified pulmonary nodules.
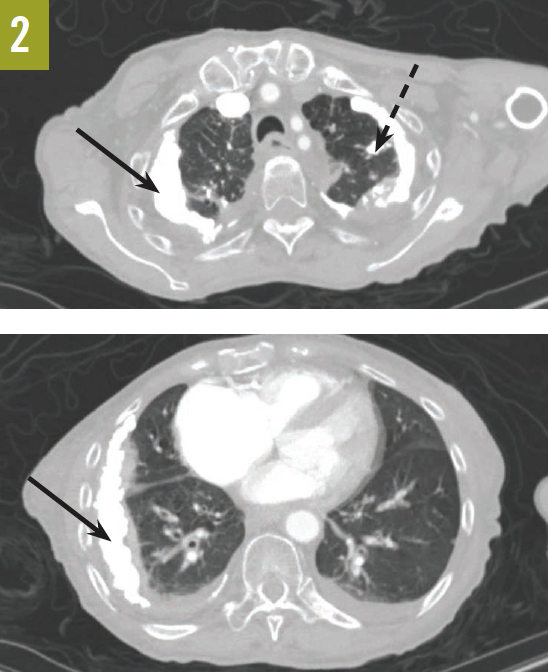
Figure 2. Chest CT scans showed extensive pleural calcifications (solid arrows) with calcified pulmonary nodules (dashed arrow).
The differential diagnosis of pleural calcification includes asbestos pleural disease with calcified pleural plaques, calcifications of prior hemothorax, empyema, exudative pleural effusions, and nonpharmacologic management of pulmonary TB. Pleural calcifications related to asbestos exposure typically arise from the parietal pleura.1 Plaques are visualized on the lateral chest wall along the sixth to ninth ribs and adjacent to the diaphragmatic surfaces of the lungs. There is relative sparing of apices and the costophrenic angles.1,2 Pleural calcifications from hemothorax, empyema, and exudative pleural effusions are localized to the areas of prior involvement.
Our patient presented with bilateral pleural calcifications mostly involving the upper lung fields. These findings were inconsistent with the pleural calcifications seen with asbestos exposure. The pattern of calcification in our patient was highly suggestive of prior pneumothorax treatment for pulmonary TB.
Before the availability of effective pharmacologic therapy, pulmonary TB was treated with “lung rest” achieved with one of the following techniques: artificial creation of pneumothorax; thoracoplasty with localized collapse of the rib cage around the tuberculous lesion; and plombage (the insertion of a foreign body into the pleural space adjacent to the site of active TB).3-5 The idea behind these measures was to reduce lung ventilation locally and close cavities that might otherwise continue to enlarge, thereby preventing the affected area of the lung from expanding, and thus “starving” the tubercle bacilli of oxygen.
Artificial pneumothorax treatment for TB was termed “collapse therapy.”4 The thinking was that if the collapse were sufficiently great, it would bring into apposition the cavitary walls, thereby hastening their closure.6 In addition, it was postulated that the contracted lung would develop a certain degree of relative ischemia, as well as a decrease in lymphatic drainage, and it was hoped that this combination of factors would result in a marked decrease in the absorption of “tuberculous toxin.”6,7
On close questioning, our patient revealed a history of pneumothorax treatment for pulmonary TB in 1939 at the age of 11, and then again in 1942. In general, pneumothorax treatment involved placing the patient on one side, with the affected hemithorax elevated. A puncture site was located between the fourth and seventh ribs in the midaxillary line. A pneumothorax needle was then introduced and advanced until the pleural cavity was reached, and a manometer showed respiratory oscillations. Initially, 50 mL of nitrogen gas would be introduced in order to avoid mediastinal organ displacement and patient discomfort. Patients would then return to the clinic every 2 to 3 days, allowing the clinician to introduce larger volumes of nitrogen gas until a desired size of pneumothorax was achieved. Because nitrogen gas was lost due to absorption, patients would return weekly in the first month, and every 7 to 14 days thereafter, for maintenance of the pneumothorax.6
Pneumothorax treatment was practiced in the late 19th and early to mid 20th centuries. Today, this previously treated population consists solely of the elderly. Health care providers should be aware of the late complications of pneumothorax treatment for TB. Since the first randomized trial of streptomycin against pulmonary TB was carried out in 1946 and 1947 by the Medical Research Council Tuberculosis Unit,8 many patients alive today never received pharmacologic therapy. Therefore, reactivation of TB due to age or other forms of immune dysfunction is of major concern in this patient population. In addition, as a further complication, the calcified pleural plaques and fibrosis produced by pneumothorax treatment can result in the development of chronic respiratory insufficiency secondary to trapped lung.
References:
- Bickle I, Gaillard F. Pleural plaque. Radiopaedia. http://radiopaedia.org/articles/pleural-plaque. Accessed November 8, 2016.
- King TE Jr. Asbestosis. UpToDate. http://www.uptodate.com/contents/asbestosis. Updated June 5, 2013. Accessed November 8, 2016.
- Reid H.Extrapleural pneumothorax in the treatment of pulmonary tuberculosis. Thorax. 1946;1(4):211-238.
- Skavlem JH. The present-day usage of pneumothorax in the treatment of pulmonary tuberculosis. Calif Med. 1950;73(6):569-572.
- Weerakkody Y, Gerstenmaier JF. Plombage. Radiopaedia. http://radiopaedia.org/articles/plombage. Accessed November 8, 2016.
- Balboni GM. The treatment of pulmonary tuberculosis by artificial pneumothorax, according to the method of Forlanini—with a report of 21 cases. Boston Med Surg J. 1912;167(22):755-761, 804-808.
- Childerhose RK. Pneumothorax treatment of tuberculosis. Radiology. 1936;27(6):741-748.
- Streptomycin treatment of pulmonary tuberculosis: a Medical Research Council investigation. Br Med J. 1948;2(4582):769-782.
NEXT: Coccidioidomycosis
Coccidioidomycosis
Authors:
John A. Goritsas, MD
University of Texas Health Science Center at San Antonio, Texas
Sayed K. Ali, MD
University of Central Florida College of Medicine, Orlando, Florida
Citation:
Goritsas JA, Ali SK. Coccidioidomycosis. Consultant. 2016;56(12):1119-1120.
A 46-year-old Hispanic woman visiting Texas from Mexico presented to the emergency department (ED) with a 3-month history of progressive shortness of breath and a cough. One week prior to her presentation, she had developed intermittent hemoptysis, prompting her visit to the ED.
History. She had a past medical history significant for mild asthma and psoriatic arthritis. She denied any fevers, chills, night sweats, or weight loss. She had been cared for regularly at a medical facility in Mexico, and her current medications included an albuterol inhaler and immunosuppressive therapy for her psoriatic arthritis, including prednisone daily and methotrexate 3 times a week.
She lived with her family in Mexico and denied ever having smoked or consumed alcohol. She also denied extensive exposure to secondhand smoke. She had been married for more than 10 years, and her daily routine involved taking care of 3 toddlers and attending to household chores.
Physical examination. On presentation, she was afebrile with a normal blood pressure. She was slightly tachypneic, with an oxygen saturation of 93% on room air. She was able to carry on a full conversation, seldom interrupted by cough. She was alert and oriented to person, time, and place. No mandibular, axillary, or femoral lymphadenopathy was appreciated.
She had a normal S1 and S2 without murmurs, rubs, or gallops. Pulmonary examination findings were significant for bilateral scattered inspiratory wheezes and bibasilar crackles, greater on the right side. Egophony sounds via auscultation were more prominent on her right side. Her abdomen was soft, nontender, and nondistended. An appendectomy scar was present on her right lower quadrant, and she had a 3-cm psoriatic plaque over the umbilicus. She had good peripheral pulses with no lower extremity edema. No joint tenderness or swelling was appreciated.
Diagnostic tests. Her white blood cell count was 8100/µL, with 6.3% eosinophils (absolute eosinophil count, 500/µL). Hemoglobin test, hematocrit test, and complete metabolic panel results were unremarkable. The erythrocyte sedimentation rate (ESR) was 62 mm/h, and the C-reactive protein level was 2.4 mg/L. Two sets of blood cultures drawn in the ED were reported as negative for pathogens. A procalcitonin level also drawn in the ED was less than 2 ng/mL.
Early morning sputum cultures drawn on different days showed normal oral flora. A Mantoux tuberculin skin test was nonreactive at 72 hours, and multiple acid-fast bacillus stains and cultures were reported as negative. QuantiFERON-TB Gold test results were also reported as negative for tuberculosis (TB). Her HIV antibody test was nonreactive, with no detectable viral load. An electrocardiogram displayed normal sinus rhythm, with no changes suggestive of ischemia.
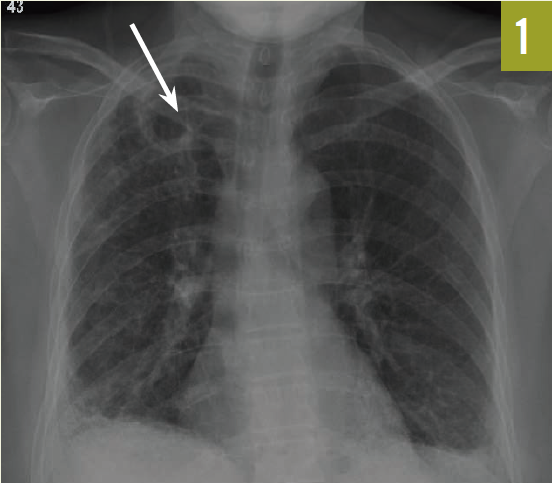
A chest radiograph revealed bilateral pulmonary fibrosis, with the right side worse than the left, with a cavitary lesion of the right apex (Figure 1, arrow). A subsequent chest computed tomography scan showed multiple right upper lobe cavitary lesions, right suprahilar lymphadenopathy, and fibrotic changes in the lung bases (Figure 2).
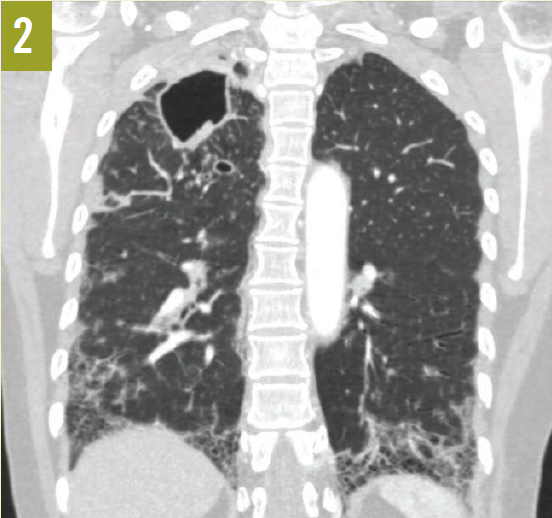
Given the patient’s right upper lobe cavitary lesion and negative TB tests, fungal serology tests for Histoplasma, Cryptococcus, Coccidioides, and Blastomyces were ordered. She underwent a bronchoscopy, which showed nonspecific chronic inflammation. Bronchoalveolar lavage showed many nucleated cells (68% eosinophils and 21% neutrophils). Calcofluor-white stains of bronchial washing were negative, but cultures grew Coccidioides immitis. A few days later, the immunoglobulin M and G serum serology tests confirmed the diagnosis of coccidioidomycosis.
Treatment. The patient was started on fluconazole, 400 mg daily, with marked improvement in her symptoms prior to discharge.
Discussion. Coccidioidomycosis (also known as valley fever) is an environmentally acquired, pulmonary fungal infection caused by inhalation of spores of Coccidioides species.1 Coccidioides is a dimorphic fungus that grows in the arid soil of the San Joaquin Valley in California, western Texas, southern Arizona, southern New Mexico, and the desert areas of northern Mexico.1,2 In addition, this fungus is also common in South American and Central American countries, including Brazil, Bolivia, Paraguay, and Argentina.3
Humans are commonly infected via the respiratory route. Outdoor activities or recreational exposure to dust greatly increases the risk of infection.1,2 It has been estimated that the incidence of coccidioidomycosis in the United States is approximately 150,000 new cases per year.4 The incidence varies with season, with the highest rate of infections in the summer, when the soil is dry and crops harvested.2
Approximately 60% of coccidioidomycosis infections are subclinical and self-limiting.1 For the reminder of cases, symptoms usually arise 1 to 3 weeks after exposure to the fungus.2 Presentation can vary from a self-limited flulike illness to pneumonia.2 Coccidioidomycosis can also have cutaneous manifestations, such as erythema nodosum or erythema marginatum.2 The majority of persons who develop symptoms experience a self-limited infection that often resolves spontaneously after a few weeks.1-4
Patients usually develop symptoms reminiscent of community-acquired pneumonia, such as fever, cough, chest discomfort, arthralgia, malaise, fatigue, and headaches. Coccidioidomycosis is virtually indistinguishable from common bacterial causes of pneumonia without the employment of specific laboratory tests.1-4 Antibiotic therapy often is initiated by the unsuspecting clinician, who is further misled by the subsequent resolution of symptoms, given the natural progression of the disease.1 To avoid this pitfall, a clinician should maintain a high index of suspicion for coccidioidomycosis, especially if the patient reports having traveled or resides in an endemic area.1
An elevated ESR and eosinophilia may be seen in patients with coccidioidomycosis but are not consistent findings.1,3 Radiographic features of coccidioidomycosis are diverse and nonspecific.3 During the acute phase of the disease, focal or diffuse infiltrates, pleural effusions, and adenopathy are frequently identified. Pulmonary nodules and cavities are present in less than 5% of cases.3 Progressive pneumonia with necrosis and cavitation is usually seen if the host’s immune system is compromised or with the use of chronic corticosteroid therapy.1 Pregnant women, especially those in the third trimester, are at higher risk for disseminated disease, but the exact reason remains unclear.2
The gold standard for diagnosing coccidioidomycosis is a positive culture or microscopic identification of the organism in clinical specimens.3 In an outpatient setting, it might be more feasible to pursue serologic testing for antibodies, which appears to be the most widely used method of diagnosing coccidioidomycosis.3
The decision to treat is based on the severity of the disease and the immune status of the host.3 Since the majority of patients with coccidioidomycosis are asymptomatic, antifungal therapy may not always be necessary. Periodic reassessment of symptoms and radiographic findings to monitor for resolution of the disease might suffice.4
Commonly prescribed therapies include oral azole antifungal agents (ketoconazole or fluconazole) dosed at 200 to 400 mg daily, usually for 3 to 6 months.4 Fluconazole is preferred for central nervous system involvement due to its good penetration of the blood-brain barrier.1 Amphotericin B is also an option, but because of its toxicity, it is reserved for severe cases.4
Regardless of a decision to treat or not, close clinical follow-up every 1 to 3 months for 1 year or longer is recommended to monitor for progression or even dissemination of the disease.4
Outcome of the case. At a 3-month follow-up visit, the patient’s shortness of breath had completely resolved with an intermittent nonproductive cough. She returned to Mexico soon thereafter, but a phone call at 6 months confirmed the complete resolution of all symptoms. We advised her to follow up with her primary care provider in Mexico for imaging studies to document the complete resolution of her lung cavity and the possible need for continued prophylaxis, given her medication-related immunodeficiency.
References:
- Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30.
- Kirkland TN, Fierer J. Coccidioidomycosis: a reemerging infectious disease. Emerg Infect Dis. 1996;2(3):192-199.
- Malo J, Luraschi-Monjagatta C, Wolk DM, Thompson R, Hage CA, Knox KS. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11(2):243-253.
- Galgiani JN, Ampel NM, Blair JE, et al; Infectious Diseases Society of America. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223.