
Peer Reviewed
Glaucoma: A Primary Care Review With a Focus on Medication Management
Authors:
Leonid Skorin Jr, DO, OD, MS; Dallas Blanco, DO; and Laura Goemann, OD
Citation:
Skorin L, Blanco D, Goemann L. Glaucoma: a primary care review with a focus on medication management. Consultant. 2017;57(6):336-341.
ABSTRACT: Glaucoma can cause progressive, irreversible vision loss if it is not promptly and properly diagnosed and treated. While the disease is typically managed by ophthalmology specialists, its wide prevalence means that primary care providers should have a fundamental understanding of glaucoma and its treatment, since the medications employed against it can have a wide array of systemic adverse effects. In addition, many common systemic medications can have adverse effects on the progression of glaucoma. This article discusses the basic principles of glaucoma, several of the most common forms of the disease, and its treatment.
KEYWORDS: Glaucoma, primary open-angle glaucoma, normal-tension glaucoma, angle-closure glaucoma
Glaucoma is the second leading cause of blindness worldwide (the first being cataracts).1,2 Early recognition and treatment of the disease is critical in the prevention of severe vision loss. Unfortunately, most forms of glaucoma follow an insidious course, and most patients are asymptomatic until significant damage to the optic nerve has occurred. In fact, vision is not typically affected until 50% of the retinal nerve fiber layer has been lost.3 Worldwide, only 10% of those who have glaucoma receive a diagnosis, and this number increases to only 50% in developed nations.4 This ubiquity merits a fundamental understanding of the nature of glaucoma by all physicians, but particularly by those involved in primary care. These physicians will be not only the initial point of contact for patients with the disease, but also the caretakers of a patient population with a multitude of other health and social issues that are impacted by the progression and treatment of glaucoma. This article aims to provide a foundation for the all-around care of patients with glaucoma by reviewing several more common variations of the disease and by discussing the relevant anatomy and physiology. It also highlights the common medications employed in the treatment of glaucoma and their adverse effects, as well as discusses medications that can exacerbate glaucoma or adversely react with glaucoma drugs.
Physiology of Glaucoma
While it is often thought of as a condition of elevated intraocular pressure (IOP), glaucoma is actually an array of diseases that results in damage to the optic nerve, particularly the nerve fiber layer of the retina.1 While increased IOP definitely plays a significant role in the disease, it is now understood that multiple factors influence the progression of glaucoma. These factors include variations in ocular anatomy, vascular health, and the pressures of the blood and cerebrospinal fluid (CSF) relative to the pressure in the eye.5 In spite of this more refined view, the monitoring and control of IOP is currently the only effective means of managing the disease.6 IOP is largely regulated by the balance of production and outflow of aqueous humor in the eye. Therefore, a fundamental understanding of the anatomy and physiology of the eye as it relates to the management of the aqueous humor is required for an understanding of glaucoma.
The aqueous humor maintains the IOP that keeps the globe structurally sound and provides nourishment to the avascular structures of the eye, including the posterior cornea, lens, and trabecular meshwork. The aqueous humor is produced by the ciliary body, located behind the iris in the posterior chamber. Production of the aqueous humor is mediated autonomically.
An increase in production occurs with parasympathetic stimulation, and a decrease occurs with sympathetic stimulation. From the posterior chamber, the aqueous humor flows through the pupil and into the anterior chamber, where it enters the trabecular meshwork in the anterior chamber angle. From the trabecular meshwork, it is taken into the canal of Schlemm and returned to the circulation through the episcleral veins. While the majority of the aqueous humor exits the eye in this fashion, 10% to 50% is removed by uveoscleral drainage through the venous circulation of the ciliary body, choroid, and sclera.7 The relative disruption of adequate outflow results in the high IOPs associated with glaucoma and the inevitable and irreversible optic neuropathy that defines the disease.
Elevated IOP exerts pressure on the optic nerve head and the blood vessels supplying the nerve. This causes ischemic damage and mechanical stress to the retinal nerve fibers as they pass through the lamina cribrosa sclerae. The ischemia and stress interfere with axoplasmic flow, interrupting the transport of nutrients, metabolic waste, and neuronal growth factors and resulting in apoptosis of these neurons.
Primary Open-Angle Glaucoma
Primary open-angle glaucoma (POAG) is the most common form of glaucoma.8 It is estimated that more than 57.5 million people worldwide have POAG, and this number is expected to rise to 76 million by 2020.9 The defining characteristics of POAG are IOPs over 21 mm Hg, glaucomatous damage to the optic nerve, and an open anterior chamber angle. The etiology of POAG is the obstruction of the trabecular meshwork by proteinaceous debris that collects over time as aqueous humor filters through it. As this debris further encumbers the trabecular meshwork, it becomes more difficult for the aqueous humor to filter through into the canal of Schlemm. This obstruction disrupts the balance between the production and the outflow of the aqueous humor, leading to chronically elevated IOPs and, eventually, damage to the optic nerve.
The development of POAG has several known risk factors. The risk of progression of glaucomatous damage to the optic nerve increases by 11% for every 1 mm Hg increase in IOP.1 Age is a strong predisposing factor, with prevalence increasing between 5 and 10 times between the fifth and eighth decades of life.1,10 Genetics also plays a large role in the development of POAG. POAG shows a predilection for Hispanic and African American populations.10 In fact, African Americans not only are more likely to develop POAG but also have an earlier progression and are more likely to present for evaluation in an advanced state.11 A family history of POAG is also a strong predisposing factor. While the inheritance of POAG is largely polygenic, 10% of cases are caused by single mutations. Two implicated genes are OPTN, which encodes the protein optineurin, and MYOC, which encodes the myocilin protein found in the trabecular meshwork.9 Several MYOC variants exist that result in production of myocilin that is aggregated within the trabecular meshwork cells rather than being transported into the meshwork, leading to dysfunction.12 Comorbidities, such as myopia, diabetes mellitus, and vasospastic disorders including migraines and Raynaud phenomenon, have also been associated with increased risk of developing POAG.10,13,14
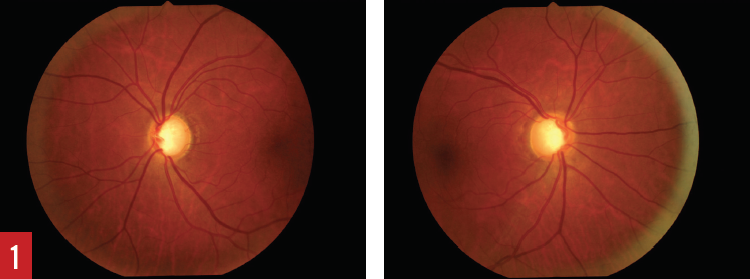
Figure 1. Deep optic cup of the left eye (left) and the right eye (right) associated with increased cup-to-disk ratio.
Most POAG patients will not self-present until the disease has already progressed significantly. This is due to the amount of extensive optic nerve damage required to produce any noticeable visual defects. Many cases are thus diagnosed during routine eye examinations and can be managed before significant damage has occurred. Widespread screening is largely impractical, causing screening to be limited to high-risk groups. Patients in these high-risk groups are individuals older than 40 years, those with an immediate family history of POAG, and those of African American or Hispanic ethnicity.10 Screening in these cases requires more than monitoring of IOPs, since there are people with ocular hypertension who never develop glaucoma, as well as people who develop glaucoma without elevated IOP (normal-tension glaucoma).6 Visual field testing and ophthalmoscopy should be part of the screening process in addition to IOP measurements. Ophthalmoscopic examination will reveal a deepening of the optic cup and associated increase in cup-to-disk ratio (Figure 1). Bayonetting of vessels and excessive collateral vessel formation may also be seen. Drance hemorrhages occur at the blood vessels adjacent to the optic nerve head (Figure 2). These hemorrhages are not specific to glaucoma, although their presence can be an indication of glaucomatous progression. Differential diagnoses to consider are vitreous detachments and diabetic or hypertensive retinopathies.15 In the absence of these conditions, progression of optic nerve damage due to glaucoma is thought to have probably occurred, and subsequent visual field defects will likely be present in the future.15 Severe glaucoma can cause very dense and progressive visual field defects (Figure 3).
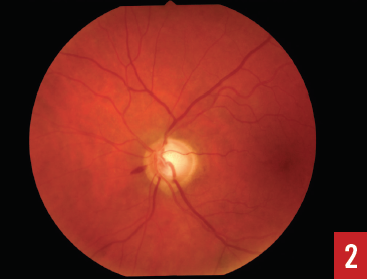
Figure 2. Disk (Drance) splinter hemorrhage.
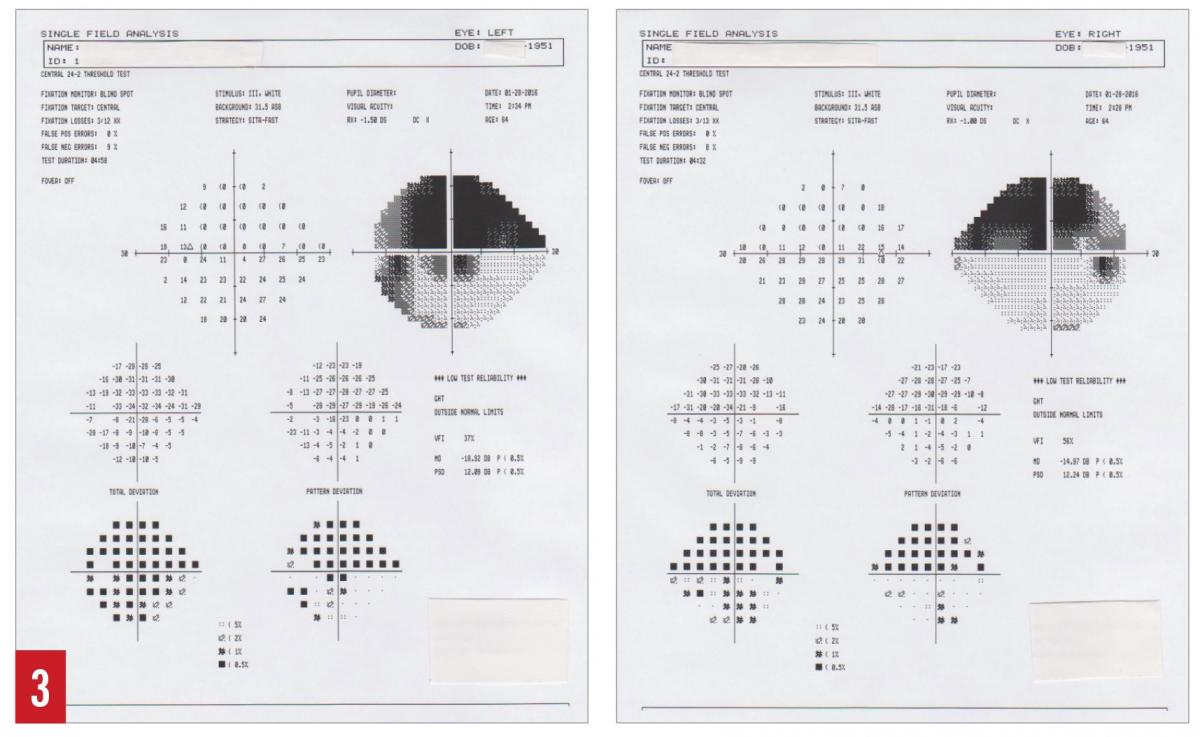
Figure 3. Superior arcuate visual field defect with inferior visual field defects of eye (right) and left eye (left) due to glaucomatous damage to optic nerve.
Treatment of POAG most often begins with ophthalmic medication. Prostaglandin analogues are considered first-line because of their effectiveness and minimal adverse effects, although topical β-blockers, α2 agonists, and carbonic anhydrase inhibitors are also all treatment mainstays. Medication choice depends on comorbidities, allergies, and tolerability, as well as individual effectiveness and cost. Lasers also may be employed as first-line or supplemental therapy. In cases that are refractory to medication or lasers, surgical treatment may be employed.
Continued on next page
Normal-Tension Glaucoma
Not all individuals with IOPs greater than 21 mm Hg develop glaucoma, while some individuals with IOP measurements consistently less than 21 mm Hg do.6 Glaucomatous optic nerve damage in patients with an IOP of less than 21 mm Hg is referred to as normal-tension glaucoma (NTG) or low-tension glaucoma (LTG). This IOP level is based on the established average range of pressures (11-21 mm Hg). There is considerable contention over whether or not this disease is merely a continuum on the spectrum of POAG.4
While the true cause of NTG is debatable, several hypotheses have been postulated to explain the glaucomatous damage that occurs in these patients with “normal” IOP levels. Anatomic variation is one hypothesis, with larger eyes and larger optic disks predisposing the optic nerve to mechanical damage from normal IOP.4
Variable corneal thickness is another anatomic concern, and the reason is 2-fold. One mechanism lies in the disruption of IOP measurement; increased corneal thickness disrupts the measurement of IOP, leading to falsely elevated readings, whereas decreased corneal thickness has the reverse effect and yields falsely low IOP readings.4 This is thought to lead to cases of individuals with POAG who appear to have normal IOPs.4 Secondly, and probably more important, a thin central corneal thickness is considered an independent risk factor for glaucoma, possibly corresponding to a thin lamina cribrosa sclerae. Conversely, an increase in central corneal thickness is thought to correspond to an increased thickness in laminal cribrosa sclerae thickness, resulting in increased stability of the optic nerve head.
Another postulated mechanism for NTG is chronic metabolic damage to the optic nerve predisposing it to mechanical damage by normal IOPs. A poorly nourished nerve is vulnerable to additional insults.6 NTG is heavily associated with abnormal vasoregulation and conditions such as migraines and Raynaud disease.14 It is also associated with systemic hypotension and exaggerated nocturnal dips in blood pressure.16,17 This has also led to the idea that glaucoma is caused not by elevated absolute IOP, but rather by the relative difference between IOP, systemic blood pressure, and CSF pressure. In cases of systemic hypotension, the ocular perfusion pressure is low, making it more challenging to nourish the optic nerve. Low CSF pressures relative to IOP have also been implicated in the pathogenesis of NTG, since the relatively high IOP pressure can cause significant mechanical stress against the optic nerve head.5,18
Treatment of NTG largely follows the same principles as treatment of POAG. Further decrease of the relatively normal IOP in these patients has been shown to prevent progression of the disease. This pattern lends further credence to the thought that the disease is merely a continuum of POAG.4 However, the initial choice of treatment may vary; the α2 agonist brimonidine is often preferred over other medications due to its presumed neuroprotective effects in addition to its IOP-lowering effect.19,20
Angle-Closure Glaucoma
Primary angle-closure glaucoma (PACG) is defined by a characteristic obstruction of the anterior chamber angle. The peripheral iris becomes closely apposed to the cornea, preventing aqueous humor from exiting through the trabecular meshwork. While PACG only accounts for one-third of glaucoma patients worldwide, it is responsible for half of all glaucoma-related blindness.4,21 It is also the predominant form of glaucoma in those of East Asian and Inuit descent.22-24
The primary mechanisms for the closure of the angle are pupillary block and nonpupillary block. Pupillary block occurs when increased pressures in the posterior chamber displace the lens forward into the iris, blocking the flow of aqueous humor through the pupil into the anterior chamber (Figure 4). This causes further pressure in the posterior chamber, causing the iris to bow forward, further closing off the anterior chamber angle.
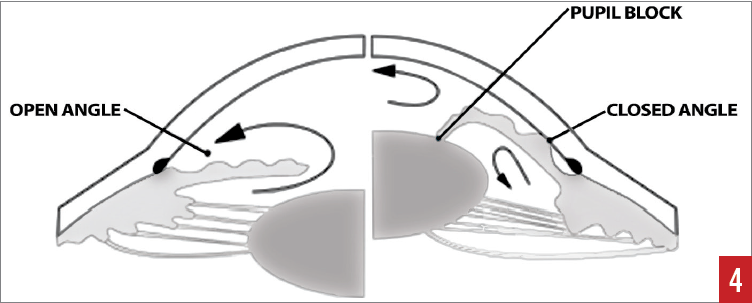
Figure 4. Aqueous flow from posterior to anterior chambers. The cross-section on the right depicts pupillary-block angle closure. Reproduced with permission from The Indian Optician, September-October 2016.
Nonpupillary block PACG is due to anatomic variations, with predisposed individuals having greater thickness in the periphery of the iris. While the majority of these patients experience a slow, progressive disease process similar to POAG, 25% will experience an attack of acute angle-closure glaucoma (AACG) which constitutes an ocular emergency.21 These patients will present with ocular pain, headache, nausea, vomiting, and blurred vision.21 IOP in these attacks can be extremely high, typically ranging from 40 to 90 mm Hg.21
Treatment for AACG includes topical cholinergic agents such as pilocarpine. These agents cause pupillary constriction and draw the iris away from the angle. Topical IOP-lowering agents such as β-blockers and systemic carbonic anhydrase inhibitors or hyperosmotic agents act rapidly to lower IOP. Laser peripheral iridotomy is performed to manage chronic PACG and to prevent future AACG events. During this procedure, the iris is perforated with a laser, allowing an alternative route of aqueous flow. Topical pilocarpine is prescribed in the AACG attack to decrease IOP and prepare the eye for laser iridotomy. In the case of AACG, identification of possible triggers is also of concern, since 33% of acute attacks are triggered by other medications.25
Glaucoma Medications
Topical prostaglandin analogues (PGAs) are widely considered first-line POAG treatment.26 PGAs lower IOP by enhancing uveoscleral outflow of the aqueous humor and providing an alternative path from the obstructed trabecular meshwork.26 PGAs used include latanoprost, travoprost, bimatoprost, and tafluprost. Several local adverse effects are common, including thickening and lengthening of the eyelashes and conjunctival hyperemia. Less common effects include a permanent darkening of the iris and periorbital fat loss.26,27 Systemic symptoms are rare, but PGAs have been known to trigger migraines and even precipitate mild upper respiratory tract symptoms.28,29
Topical β-blockers are highly effective at lowering IOP by decreasing the production of aqueous humor by the ciliary body. These drugs include timolol, betaxolol, and levobunolol. Their usefulness is diminished because of their adverse effects. Despite their administration as topical eye drops, β-blockers can have dramatic systemic effects. Cardiovascular effects are prominent and include bradycardia, heart block, and hypotension, which can lead to falls. Peripheral vasoconstriction can also be problematic in those who have Raynaud syndrome. Bronchospasm is another systemic effect to consider, particularly in patients with a history of asthma or chronic obstructive pulmonary disease (COPD).30 A myriad of other systemic adverse effects are associated with these drugs, including sleep disorders, reduced exercise tolerance, decreased libido, erectile dysfunction, confusion, depression, and dyslipidemia.
Topical α2 agonists act on the ciliary body to reduce aqueous humor production as well as promote uveoscleral aqueous outflow.31 They also have a subtle neuroprotective effect, directly reinforcing the optic nerve against glaucomatous damage.19,20 Brimonidine and apraclonidine are examples of agents in this class. Worsening of vascular insufficiency is a possible adverse effect of these medications. Moreover, α2 agonists are capable of crossing the blood-brain barrier and causing central nervous system depression and hypotension, with particularly severe effects in children.32 These ophthalmic eye drops are contraindicated in patients younger than 2 years,32 and even adults may notice daytime sleepiness. They also should not be given alongside monoamine oxidase inhibitors (MAOIs), because this combination can precipitate a hypertensive crisis.
Topical carbonic anhydrase inhibitors decrease the production of aqueous humor, given that the formation of bicarbonate is key to the ciliary body in establishing an osmotic gradient to pull fluid into the eye. Dorzolamide and brinzolamide are among the agents in this class. These drugs are structurally similar to sulfa antibiotics, and a sulfa allergy is a relative contraindication to their use due to potential cross-reactivity; however, an actual reaction is very rare. Systemic carbonic anhydrase inhibitors are employed to decrease IOP spikes in an acute setting. Acetazolamide and methazolamide are in this medication class. In addition to the relative contraindication in patients with a sulfa allergy, they can cause significant systemic adverse effects, including paresthesias, hypokalemia, renal stones (rare), and general malaise with flulike symptoms. Rare but serious adverse effects can occur, such as Stevens-Johnson syndrome, bone marrow suppression, and aplastic anemia.33,34
Osmotic agents are used only in emergency departments and operating rooms. These include mannitol and isosorbide, which can be employed in a similar fashion to systemic carbonic anhydrase inhibitors and work by drawing fluid out of the eye and lowering acutely elevated IOPs. Care must be taken in patients with congestive heart failure, since these drugs will increase the cardiovascular load.
Miotics are used to treat both POAG and AACG. They act by causing pupillary constriction, thinning the peripheral iris, and opening the anterior chamber angle. Systemic adverse effects are rare but may include confusion, bradycardia, bronchospasm, and urinary frequency. Pilocarpine is the most commonly used member of this class, although it is not very frequently used now given the availability of other agents. Adverse effects include decreased night vision and increased risk of cataract formation. In addition, miotics must be used at least 3 times a day for therapeutic effect, making them less convenient for many patients.
Medicines That Exacerbate Glaucoma
Anticholinergic medications used systemically are contraindicated in patients with some forms of glaucoma, primarily untreated narrow-angle glaucoma.35 These medications include ipratropium used in COPD and tolterodine used in overactive bladder. Anticholinergics cause pupillary dilation, which can close off the anterior chamber angle in patients with structurally narrow angles. They do not have any significant negative impact on POAG. These medications are contraindicated in patients with AACG but are safe to use in patients who have been treated with laser iridotomy.
Many drugs possess potent anticholinergic effects, and their use in the case of AACG should also be carefully considered. Tricyclic antidepressants, selective serotonin-reuptake inhibitors, MAOIs, antipsychotics, and antihistamines all commonly have anticholinergic effects. Sympathomimetics, similarly to anticholinergics, cause pupillary dilation and can exacerbate AACG. Epinephrine used for severe asthma or in cases of anaphylaxis, and ephedrine, which is found in a variety of cold medications, have adrenergic effects in the eye.36
Corticosteroids can raise the IOP in a portion of the population known as steroid responders.37 This trait is highly inheritable and correlates with POAG; up to 70% of offspring of patients with POAG are steroid responders.38 While systemic steroids can cause this effect, it is much more pronounced and common in steroids that are administered as eye drops or nasal sprays. However, even topical steroid creams can cause an increase in IOP in susceptible patients. Patients with POAG, as well as their immediate relatives, are more likely to be steroid responders, and steroid responders are more susceptible to developing POAG. The use of steroids in patients with or at risk for POAG should be accompanied by careful observation.39
Even when given systemically, β-blockers also lower IOP. They may also mask the potential for an individual to develop glaucoma, and discontinuing or even titrating systemic β-blockers may cause significant increases in IOP.40 These drugs have also been implicated in the development of NTG, particularly by causing nighttime hypotension when taken immediately before bed. It is thus recommended that patients with glaucoma, especially NTG/LTG, take systemic hypotensive medications in the morning to minimize systemic nighttime hypotension and its possible negative effect on optic nerve blood flow.
Leonid Skorin Jr, DO, OD, MS, is an ophthalmologist at the Mayo Clinic Health System in Albert Lea, Minnesota.
Dallas Blanco, DO, is a recent graduate of William Carey University College of Osteopathic Medicine in Hattiesburg, Mississippi.
Laura Goemann, OD, is a recente graduate of Pacific University College of Optometry in Forest Grove, Oregon.
REFERENCES:
- Bowling B. Glaucoma. In: Bowling B. Kanski’s Clinical Ophthalmology: A Systematic Approach. 8th ed. Philadelphia, PA: Elsevier; 2016:305-395.
- Yokoyama Y, Maruyama K, Konno H, et al. Characteristics of patients with primary open angle glaucoma and normal tension glaucoma at a university hospital: a cross-sectional retrospective study. BMC Res Notes. 2015;8:360.
- Quigley HA, Addicks EM, Green WR. Optic nerve damage in human glaucoma: III. Qualitative correlation of nerve fiber loss and visual field defect in glaucoma, ischemic neuropathy, papilledema, and toxic neuropathy. Arch Ophthalmol. 1982;100(1):135-146.
- Quigley HA. Glaucoma. Lancet. 2011;377(9774):1367-1377.
- Berdahl JP, Allingham RR, Johnson DH. Cerebrospinal fluid pressure is decreased in primary open-angle glaucoma. Ophthalmology. 2008;115(5):763-768.
- Cohen A, Wong SH, Patel S, Tsai JC. Endoscopic cyclophotocoagulation for the treatment of glaucoma. Surv Ophthalmol. 2017;62(3):357-365.
- Toris CB, Yablonski ME, Wang Y-L, Camras CB. Aqueous humor dynamics in the aging human eye. Am J Ophthalmol. 1999;127(4):407-412.
- Souzeau E, Burdon KP, Ridge B, Dubowsky A, Ruddle JB, Craig JE. A novel de novo Myocilin variant in a patient with sporadic juvenile open angle glaucoma. BMC Med Genet. 2016;17:30-34.
- Abu-Amero K, Kondkar AA, Chalam KV. An updated review on the genetics of primary open angle glaucoma. Int J Mol Sci. 2015;16(12):28886-28911.
- Bennet GR, Zaman F. Primary open-angle glaucoma (POAG). In: Onofrey BE, Skorin L Jr, Holdeman NR, eds. Ocular Therapeutics Handbook: A Clinical Manual. 3rd ed. Philadelphia, PA: Wolters Kluwer Lippincott Williams & Wilkins; 2011:435-439.
- AGIS Investigators. The Advanced Glaucoma Intervention Study (AGIS): 9. Comparison of glaucoma outcomes in black and white patients within treatment groups. Am J Ophthalmol. 2001;132(3):311-320.
- Hill SE, Donegan RK, Lieberman RL. The glaucoma-associated olfactomedin domain of myocilin forms polymorphic fibrils that are constrained by partial unfolding and peptide sequence. J Mol Biol. 2014;426(4):921-935.
- Lin H-C, Chien C-W, Hu C-C, Ho J-D. Comparison of comorbid conditions between open-angle glaucoma patients and a control cohort: a case-control study. Ophthalmology. 2010;117(11):2088-2095.
- Nguyen BN, Lek JJ, Vingrys AJ, McKendrick AM. Clinical impact of migraine for the management of glaucoma patients. Prog Retin Eye Res. 2016;51:107-124.
- Liebmann JM. Finding and responding to disc hemorrhages. Rev Ophthalmol. https://www.reviewofophthalmology.com/article/finding-and-responding-to-disc-hemorrhages. Published April 22, 2010. Accessed May 8, 2017.
- Charlson ME, de Moraes CG, Link A, et al. Nocturnal systemic hypotension increases the risk of glaucoma progression. Ophthalmology. 2014;121(10):2004-2012.
- Graham SL, Drance SM. Nocturnal hypotension: role in glaucoma progression. Surv Ophthalmol. 1999;43(suppl 1):S10-S16.
- Burgoyne CF, Downs JC, Bellezza AJ, Suh J-KF, Hart RT. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog Retin Eye Res. 2005;24(1):39-73.
- Kitaoka Y, Kohima K, Munemasa Y, Sase K, Takagi H. Axonal protection by brimonidine with modulation of p62 expression in TNF-induced optic nerve degeneration. Graefes Arch Clin Exp Ophthalmol. 2015;253(8):1291-1296.
- Beltramo E, Lopatina T, Mazzeo A, et al. Effects of the neuroprotective drugs somatostatin and brimonidine on retinal cell models of diabetic retinopathy. Acta Diabetol. 2016;53(6):957-964.
- Patel N, Gonzalo O, Mancera N, Patel H. Ocular emergencies in primary care: diagnosis, treatment, and referral. Consultant. 2016;56(10):892-896.
- Bourne RRA, Sørensen KE, Klauber A, Foster PJ, Johnson GJ, Alsbirk PH. Glaucoma in East Greenlandic Inuit—a population survey in Ittoqqortoormiit (Scoresbysund). Acta Ophthalmol Scand. 2001;79(5):462-467.
- Shastry BS. Genetic susceptibility to primary angle closure glaucoma (PACG). Discov Med. 2013;15(80):17-22.
- Chan EW, Li X, Tham Y-C, et al. Glaucoma in Asia: regional prevalence variations and future projections. Br J Ophthalmol. 2016;100(1):78-85.
- Lachkar Y, Bouassida W. Drug-induced acute angle closure glaucoma. Curr Opin Ophthalmol. 2007;18(2):129-133.
- Alm A, Grierson I, Shields MB. Side effects associated with prostaglandin analog therapy. Surv Ophthalmol. 2008;53(suppl 1):S93-S105.
- Jayaprakasam A, Ghazi-Nouri S. Periorbital fat atrophy—an unfamiliar side effect of prostaglandin analogues. Orbit. 2010;29(6):357-359.
- Gruener AM, Griffiths MFP. Increased systemic side effects of prostaglandin analogue eye drops in patients with palatal defects. Br J Oral Maxillofac Surg. 2012;50(3):e45.
- Weston BC. Migraine headache associated with latanoprost. Arch Ophthalmol. 2001;119(2):300-301.
- Kaiserman I, Fendyur A, Vinker S. Topical beta blockers in asthmatic patients—is it safe? Curr Eye Res. 2009;34(7):517-522.
- Toris CB, Gleason ML, Camras CB, Yablonski ME. Effects of brimonidine on aqueous humor dynamics in human eyes. Arch Ophthalmol. 1995;113(12):1514-1517.
- Coppens G, Stalmans I, Zeyen T, Casteels I. The safety and efficacy of glaucoma medication in the pediatric population. J Pediatr Ophthalmol Strabismus. 2009;46(1):12-18.
- Munshi V, Ahluwalia H. Erythema multiforme after use of topical dorzolamide. J Ocul Pharmacol Ther. 2008;24(1):91-93.
- Keisu M, Wilhelm B-E, Öst Å, Mortimer Ö. Acetazolamide-associated aplastic anemia. J Intern Med. 1990;228(6):627-632.
- Eskandar OS, Eckford SD, Whittaker KW. Treatment of overactive bladder (OAB) with anti-cholinergic drugs and the risk of glaucoma. J Obstet Gynaecol. 2005;25(5):419-421.
- Tripathi RC, Tripathi BJ, Haggerty C. Drug-induced glaucomas: mechanism and management. Drug Saf. 2003;26(11):749-767.
- Kersey JP, Broadway DC. Corticosteroid-induced glaucoma: a review of the literature. Eye (Lond). 2006;20(4):407-416.
- Bartlett JD, Woolley TW, Adams CW. Identification of high intraocular pressure responders to topical ophthalmic corticosteroids. J Ocul Pharmacol. 1993;9(1):35-45.
- Clark AF, Morrison JC. Steroid-induced glaucoma. In: Morrison JC, Pollack IP, eds. Glaucoma: Science and Practice. New York, NY: Theime Medical Publishers; 2003:197-206.
- Khawaja AP, Chan MPY, Broadway DC, et al. Systemic medication and intraocular pressure in a British population: the EPIC-Norfolk Eye Study. Ophthalmology. 2014;121(8):1501-1507.