
Common Tragedies Of Lax Joint Syndromes: Broken Hearts, Fallen Men, And Loose Women
ABSTRACT: Joint laxity syndromes can cause many nonspecific and variable symptoms, even among patients with the same condition, making diagnosis difficult. Many patients see numerous specialists before receiving a proper diagnosis, as many healthcare providers are only familiar with the more extreme forms of the disease (eg, Marfan syndrome, osteogenesis imperfecta) despite milder variants being more prevalent. This article provides an overview of the spectrum of joint laxity syndromes, examining symptoms, assessment, and workup of patients suspected to have such conditions. It also provides an overview of the role of genetic testing and of the preventive and therapeutic strategies physicians can use to improve the care of their patients.
At least 10% of the general population has some degree of joint laxity, with a tenth of these individuals experiencing pain and disability that can be ameliorated by the astute physician.1-5 Patients with joint laxity may have symptoms ranging from benign hypermobility traits2-4 that pose minimal risks (eg, arthralgia, minor injuries) to extreme diseases that can cause early death (eg, Marfan syndrome5 or Ehlers-Danlos syndrome [EDS] type IV1,6.7). When more serious signs and symptoms accompany hypermobility, patients are classified as having connective tissue dysplasia (CTD). The most common CTD is EDS, which can cause symptoms ranging from joint hypermobility and fragile skin to rupture of the blood vessels, intestines, and other organs. Patients with EDS and other CTDs often present with a variety of symptoms that affect many body systems.7-10
In addition to musculoskeletal pain or injury, patients may have migraines, menorrhagia, fragile skin with unusual healing, and/or fragile vessels with easy bruising. Pooling of blood in the distensible vessels of the lower limbs and pelvis can cause decreased cephalic flow with adrenergic stimulation and autonomic imbalance (dysautonomia). Sympathetic imbalance can lead to irritable bowel syndrome (IBS), causing reflux, bloating, constipation, and a sensitive stomach. It can also lead to postural orthostatic tachycardia syndrome (POTS), causing disabling dizziness, fainting, and intermittent fatigue.10 In addition to physical ailments, patients with CTD may experience mental status changes, such as anxiety, depression, and confusion (often referred to as brain fog).7-10
Recognition of the heterogeneous spectrum of CTDs has transferred the diagnosis from the narrow aisles of genetics into the main theatre of medicine. As such, emphasis on physician skills has been restored, as timely clinical diagnosis is paramount to ensuring proper treatment and improving quality of life. To identify EDS and other CTDs, joint laxity should be routinely screened for in clinical practice. Currently, too many patients are written off as hypochondriacs or malingerers as they go from specialist to specialist with a barrage of symptoms and inaccurate diagnoses. Prompt recognition of CTDs validates patients’ concerns and improves their quality of life, enabling proper steps to be taken to ameliorate their symptoms and protect them from injury, such as nutrition therapy, physical therapy, cognitive-behavioral therapy, and proper medicines (eg, beta-blockers to moderate tachycardia and slow vascular dissection).5,6 8-13
This article provides an overview of CTDs, focusing on identification of milder variants—much more likely to be missed. By juxtaposing 2 cases with milder disease with 1 demonstrating an extreme form of CTD (ie, Marfan syndrome), clinicians can gain familiarity with the characteristic pattern of symptoms observed in patients with CTDs, along with a clearer definition of hyperelastic skin and joints. Reviewing genetic testing and strategies for preventing and treating injuries and other CTD symptoms can prove to be highly valuable across the board.
CASE PRESENTATIONS
Although milder forms of CTD are more common, they are not as easily recognized as severe forms of the disease (eg, Marfan syndrome, osteogenesis imperfecta) because their signs and symptoms involve many body systems and are often nonspecific. The 3 case presentations that follow provide a sample of the wide spectrum of CTDs that may be encountered in clinical practice.
Case 1
A 21-year-old female athlete and honors student presented to a cardiologist after disabling dizziness, fainting, and fatigue interfered with her ability to drive, requiring her to take a leave of absence from college. The patient had been active in soccer but was sidelined by multiple sprains, several knee surgeries, and 3 limb fractures. She recognized early on that she was double-jointed, showing off maneuvers for friends, and sometimes experienced popping of multiple joints and spontaneous dislocation of her shoulders and hips. A physical examination at school revealed mild scoliosis. During her current presentation, a review of her family history revealed that 1 of her 2 sisters had similar symptoms and also sustained multiple joint injuries, but no abnormalities were noted for the patient’s parents.
The patient had an extensive medical history, with many body systems affected. She had seen 8 subspecialists before her current presentation, receiving diagnoses of rheumatoid arthritis (serology negative), fibromyalgia, vertigo, arrhythmia, Crohn’s disease, anxiety, and depression. The findings from a review of systems revealed the following:
•Autonomic—Panic attacks and frequent palpitation episodes as a teen; heat sensitivity leading to avoidance of hot showers and her friends calling her “tomato-face” after exercise, love of salt to the point of eating it from the shaker.
•Bones/Joints—Double-jointed; popping of multiple joints; spontaneous dislocation of shoulders and hips; mild scoliosis; daytime knee and ankle pain as a child; back pain as a teenager; multiple sprains, knee surgeries, and limb fractures.
•Bowel—Infantile colic with later bloating and constipation (first diagnosed as Crohn’s disease and then as IBS).
•Cognitive—Persistent fatigue after a mononucleosis-like illness; memory loss and poor focus, which was often referred to as brain fog.
•Eyes/Ears—Glasses for myopia since age 5; fainting spells precipitated by tunnel vision; tinnitus.
•Gynecologic—Irregular and heavy menses requiring moderation with birth control.
•Mouth—Dental crowding necessitating 3 courses of orthodontic treatment; temporomandibular joint disorder with jaw catching and pain.
•Neurologic—Migraines since childhood and occipital pain with radiation down her neck; frequent dizziness upon standing, especially when getting up in the morning; numbness and tingling of the extremities.
•Psychiatric—Anxiety and depression ineffectively treated with psychotropic medications.

Figure 1. Maneuvers of the Beighton hypermobility scale, excluding the knee hyperextension maneuver. Passive dorsiflexion and hyperextension of the fifth metacarpophalangeal joint beyond 90°(A). Passive apposition of the thumb to the flexor aspect of the forearm (B). Passive hyperextension of the elbow beyond 10° (C). Active forward flexion of the trunk with the knees fully extended so that the palms of the hands rest flat on the floor (D).
Physical examination revealed a normal body build, with a height and weight in the 75th and 50th percentiles, respectively, for her age. Upon examining her head, eyes, ears, nose, and throat, her face appeared neither long nor aged but she did have the high palate often seen with CTD. A cardiac examination, including an echocardiogram, was normal. The patient demonstrated poor balance on tandem walk. Upon performing a series of maneuvers to assess her hypermobility, she was given a Beighton hypermobility score of 7/9 (Figures 1A-1D), but she had a negative Walker-Murdoch sign (ie, ability to overlap the little finger and thumb around the wrist, which can indicate arachnodactyly or abnormally long and slender fingers).
An examination of her back revealed levoscoliosis less than 10º with lumbar lordosis. Her skin had a soft texture and bruised easily. There was moderate hyperelasticity, as shown by the ability to raise a 1-inch fold of skin on her forearm (Figure 2). She also had multiple striae and 2 white-surfaced (“cigarette-paper”) surgical scars over her knees (Figure 3). Other significant findings included a positive tilt-table test, elevated catecholamine levels, and low vitamin D levels. Whole exome sequencing (WES)14 revealed that the patient and her mother had a collagen V gene mutation. A diagnosis of EDS type 1 was made. Her cardiologist advised nutritional treatment of POTS as described in the text, and she felt much better.
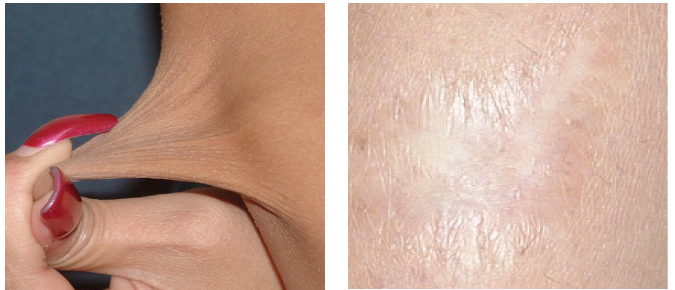
Figure 2. A 1-inch fold of raised skin on the patient’s forearm, indicating moderate hyperelasticity.
Figure 3. A white-surfaced scar, also known as a cigarette-paper scar.
Case 2
A 40-year-old woman presented to her primary care physician with a history of occasional sudden falls when walking. During these episodes, she experienced a brief loss of consciousness but recovered in time to protect herself from severe injury. She thought that her knees were giving out and wanted an evaluation. The patient’s family history was significant for a mother with increased flexibility, arthritis, and mitral valve prolapse. The patient’s medical history included the following:
•Bowel—Significant gastroesophageal reflux disease and failure to thrive as a child, with later diagnosis of Crohn’s disease requiring medication.
•Heart—Mitral valve prolapse, which was recognized after an episode of endocarditis from untreated streptococcal pharyngitis.
•Joints—Increased flexibility/clumsiness as a child with popping joints and occasional joint pain.
•Skin—Easy bruising with a few striae and white-surfaced scars.
On physical examination, the patient’s height was 5 feet 2 inches and she had a medium build. Her Beighton hypermobility score was 7/9. She had soft skin with one white-surfaced scar, minimal hyperelasticity, and no increased striae. Laboratory evaluation revealed low vitamin D levels and mild anemia.
A diagnosis of EDS type I was made based on the clinical findings and a review of the patient’s history. Fluid and salt therapy addressed her POTS and prevented new falling spells. Genetic testing was not pursued since she did not have good insurance coverage and the clinical diagnosis was sufficient for management
Case 3
During a routine sports physical a 17-year-old boy was noted to be tall and thin (>97th and 50th percentile, respectively, for age). He had pectus carinatum that became more dramatic with age (Figure 4) and reported early joint popping and pain. He was referred to cardiology, orthopedics,ophthalmology, and genetics for a suspected diagnosis of Marfan syndrome. Cardiology documented an aortic root diameter well above the 97th percentile for his age, and orthopedics documented kyphoscoliosis (Figure 5). Ophthalmology showed bilateral upward dislocation of the lens (Figure 6). Genetics noted myopia, arachnodactyly, lax skin, and mild joint hypermobility with a Beighton score of 4/9. Whole exome sequencing demonstrated a fibrillin-1 gene mutation, which is diagnostic of Marfan syndrome. His parents did not have the mutation.
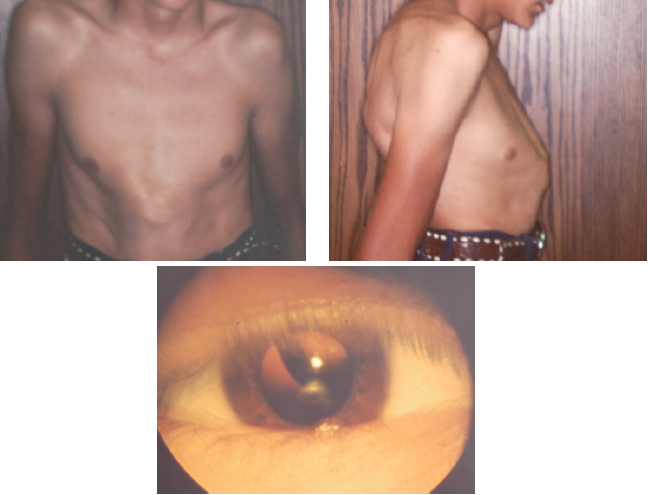
Figure 4. Patient’s pectus carinatum, which worsened with age.
Figure 5. Side profile showing the patient’s kyphoscoliosis and pectus carinatum.
Figure 6. Upward lens dislocation, as might be seen in Marfan syndrome.
The patient’s family history was unremarkable, suggesting he had a new mutation. This indicated that his siblings had minimal risks of also having the disorder, but he had a 50% risk of transmitting the disease to his future offspring. The patient received losartan, an older antihypertensive medication, to slow his aortic root dilatation. This agent has been shown to act on the fibrillin-transforming growth factor-beta pathway.8 His cardiologist forbade participation in contact sports and he was instructed to stay in touch with his joints and avoid activities that caused pain or injury (jogging is worst, swimming best). Although males are less likely to have POTS, he was educated about regular fluid, salt, and vitamin D intake to prevent chronic fatigue or tachycardia.
Examining the Cases
All 3 case patients have joint laxity syndromes, with the case 1 and 2 patients having milder disease best classified as type I EDS; the many novel genes being found in EDS patients argue for an EDS spectrum rather than clinically distinguishable types. 1-4 Case 3 is an example of severe CTD diseases that are more easily recognized by physicians and genetic specialists, whether Marfan syndrome5 with vascular dilatation or dissection, EDS IV with GI and vascular ruptures, or osteogenesis imperfect with recurring fractures and blue-gray sclera. All 3 case patients exhibited early symptoms of flexibility that included joint popping, arthralgia, and being “double-jointed.” The significance of such symptoms is often overlooked by patients because they assume that everyone has the same abilities or by physicians because they often do not assess hypermobility.11-13
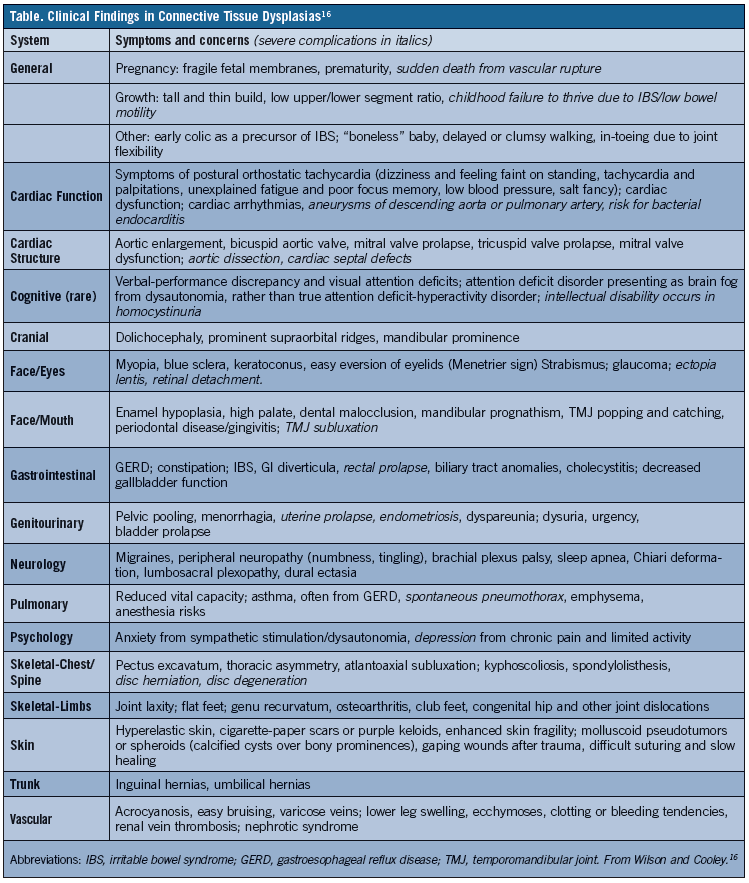
Another problem with CTD diagnosis is that flexibility symptoms may not be correlated with symptoms affecting other parts of the body. The case 1 and 2 patients had IBS, which is often misdiagnosed as Crohn’s disease or celiac disease. They also had POTS, which causes dizziness and faintness on standing that patients often address by steadying themselves after standing. Other frequent findings are chronic fatigue and poor focus or brain fog, which tend to manifest after exposure to an environmental trigger, such as trauma, infectious illnesses like mononucleosis (as occurred with the case 1 patient), and a first pregnancy in women. Therefore, when joint and/or skin laxity is identified, physicians should evaluate for other signs and symptoms that can be indicative of a joint laxity syndrome, such as those outlined in the Table.11-13
When a joint laxity syndrome is properly diagnosed, an appropriate subspecialty referral can be made. Had the condition been diagnosed earlier in the case 1 patient, she would have been spared from multiple unproductive subspecialty visits. The clinical diagnosis was sufficient to provide appropriate interventions and closure for cases 1 and 2, and to guide specific gene testing to diagnose Marfan syndrome in case 3.
DEFINING HYPERELASTIC SKIN AND JOINTS
Although historical and circus “elastic men” have been known for centuries, dermatologists Ehlers and Danlos clinically described hyperelastic skin and joints in the early 1900s.1 Diverse terms for this condition were unified as Ehlers-Danlos syndrome in the 1930s. In 1972, medical geneticist McKusick classified the syndrome as 7 distinct types.1,6 In the medical literature, EDS types I to VII with subgroups are often described, but lay groups like the Ehlers-Danlos National Foundation15 simplify this classification by separating out 3 main types as follows: classical, which is characterized as skin findings with minimal joint laxity; vascular, which is associated with vascular and bowel ruptures; and hypermobile, which is characterized by hypermobility and multisystem disease. Added to these are benign hypermobility diagnoses on the milder end of the spectrum and uncommon more severe diseases on the severe end (eg, Marfan syndrome, Stickler syndrome, osteogenesis imperfecta, and pseudoxanthoma elasticum). Because most of these disorders include some joint and/or skin elasticity in affected patients, these findings serve as proxies for the many disorders within the CTD spectrum.1
Hyperelastic skin can be demonstrated by a soft doughy texture or being able to pinch a fold of skin larger than 1 inch on the forearm (Figure 2), the exception being in patients 65 years and older. Because these measures can be subjective and influenced by a variety of factors, such as age, a more useful measure might be skin fragility. Indicators of skin fragility include dewlaps (ie, folds of loose skin hanging from the neck), excess wrinkling, aged facial appearance, easy bruising, papyraceous scars (Figure 3), keloids, hyperkeratotic plaques (eg, molluscoid pseudotumors over elbows and knees), spheroids (ie, shot-like nodules over bony prominences), and lavender macules over old insect bites. Patients with EDS type IV have a seemingly contradictory finding of tight skin around their lower face; however, these patients have thin skin that reveals the venous patterns underlying their skin, which is also seen with many CTDs.6
Assessment of joint hypermobility was made somewhat objective with the introduction of the Beighton score. This scoring system allocates one point for each side that the following maneuvers are executed on, with a maximum score of 9 points: bilateral retroflexion of the fifth finger on the right and left hand beyond 90° (Figure 1A), bending of the left and right wrist to touch the thumb to the forearm (Figure 1B), hyperextension of the left and right elbows and the left and right knees beyond 10° (Figure 1C), and touching both palms to the floor (Figure 1D). Above-average hypermobility requires a minimum Beighton score of 4 out of 9, but the average varies by age, sex, and ethnicity, with women having more skin and joint elasticity at all ages. Maneuvers that are not part of the Beighton score but that can be used to further evaluate joint hypermobility include neck rotation beyond 90°, neck side flexion beyond 50°, hip abduction beyond 90°, dorsal flexion of the distal thumb or toe beyond 90°, wrapping hand around the back to touch the umbilicus, clasping hands behind the back with one arm over the shoulder, easy eversion of upper eyelid (Ménétrier’s sign), and/or touching the nose with the tongue (Gorlin sign). The Walker-Murdoch sign (easy overlap of thumb and little finger around wrist) indicates hypermobility and arachnodactyly, which is suggestive of Marfan syndrome when accompanied by an increased arm span- to-height ratio or decreased upper-to-lower body segment ratio.5
It has been estimated that 6% to 30% of male patients and 15% to 50% of female patients, depending on age, would demonstrate some positive findings of hyperelasticity were skin and joint hyperelasticity assessments to become a routine part of clinical practice.3,4 Once any positive findings are identified, a review for clinical significance is imperative. This requires a comprehensive review of the patient’s history, including whether he or she participates in any activities requiring flexibility (eg, gymnastics, cheer, dance) and the frequency of pain, sprains, dislocations, tendon ruptures, or fractures. Arthralgia is the most frequently reported type of pain, but assessment for other pain types can be helpful, such as headaches and pain of the articular limb, muscular limb (myofascial and fibromyalgia), neuropathic limb (numbness, tingling, chronic regional pain syndromes), back/neck (with frequent disc herniation or displacement), abdominal (due to IBS), pelvic (congestion, menorrhagia, dyspareunia, endometriosis).8-13
When joint hypermobility goes beyond trait to produce pain and injury, and a careful review of the patient’s expanded history identifies any of the signs and symptoms listed in the Table, the likelihood of a CTD diagnosis is strengthened. A study of life experiences in 466 individuals with EDS using an online survey showed that 99% experienced joint pain before age 18 years and 99% had hypermobility, these joint-related symptoms often accompanied by other conditions, including chronic fatigue in 82%, anxiety in 73%, depression in 69%, and fibromyalgia in 42%.8-13 As many as one-third of respondents had 15 to 25 symptoms arising from their EDS.13
GENETIC TESTING FOR JOINT LAXITY SYNDROMES
The EDS/CTD spectrum ranges from severe genetic disorders associated with single gene changes to milder, multifactorial disorders that stem from combinations of genetic and environmental factors. Genetic testing can focus on single genes like fibrillin-1 associated with Marfan syndrome in patients with extreme symptoms like case 3, but must examine many genes in patients with broader symptoms like those of cases 1 and 2. (WES)14 that analyzes the coding regions (exons) of all 23,000 genes is now the test of choice for milder patients, its $10,000 cost justified by much greater sensitivity than single gene tests that cost between $1000 and $3000 each, even greater than sequencing panels of 15 to 30 genes related to CTD (~$4000) because many new genes related to CTD are being discovered by WES.6,17
Surprisingly, WES is better covered by insurance companies with over half the author’s patients having zero out-of-pocket costs, and it has the additional advantage of screening genes like those for breast-ovarian cancer (BRCA), undertaken as “incidental findings” that are separately consented. Physicians and patients can find information about particular CTD conditions18 or the laboratories conducting various genetic tests19 by searching on the appropriate websites.
Advantages of gene testing beyond that of establishing a clinical diagnosis should be weighed carefully, as therapy and preventive healthcare measure can be guided based on a patient’s history and physical examination findings. Most CTD, including extreme conditions like EDS type IV, Marfan syndrome, and osteogenesis imperfect, exhibit autosomal dominant inheritance, with affected individuals having a 50% risk of transmitting the disease to their offspring with each pregnancy.
When the parents of an affected child do not have any recognized CTD mutations, it can be assumed that the affected child has a new mutation (with the rare exception of germline mosaicism where a parent has several gametes with the mutation and has a 1% to 2% rather than zero risk for the next child to be affected). A confounding factor in estimating transmission risk is variable expression, where people with the same mutation, even in the same family, have very different symptoms that range from minimal (incomplete penetrance) to full-blown CTD. Genetic testing can reveal the minimally affected parent who actually has a CTD mutation with 50% rather than low transmission risk, but it can only document the presence of a mutation (eg, in an embryo using preimplantation genetic diagnosis,20 or in a fetus using amniocentesis), not the severity of disease that individual will manifest.
The more common, milder presentations of CTD can result from single gene mutations that produce milder symptoms, like that in collagen V for case 1, but can also involve mutations in several genes (multifactorial determination) as is increasingly found by WES. Because several gene mutations will have a lower chance to be transmitted together from parent to child, multifactorial CTDs have only 10% to 20% risks of transmission from parent to child. Thus prediction of transmission risks (recurrence risks) for patients with CTD is complex, even after delineation of mutations by genetic testing. Patients with milder CTD have transmission risks from 10% to 50% with each pregnancy depending on the operation of single gene (autosomal dominant) or multigenic (multifactorial) inheritance, and they can be reassured that the gene(s) will not transform into those for an extreme disorder like Marfan syndrome or EDS type IV.
Genetic testing can exclude the presence of an extreme CTD disorder and discriminate between a 10% (multigenic) or 50% (single gene) transmission risk, with severity of affected offspring unpredictable but at least limited to those of milder CTD. This genetic counseling would apply to case 1 with a collagen V mutation, a 50% transmission risk with variable but not extreme disease symptoms in her offspring.
Case 3 with Marfan syndrome would receive the same genetic counseling with the proviso that offspring symptoms could include potentially lethal aortic dilatation. The case 2 patient would have to undergo gene (DNA) testing to determine her risk of transmission, allowing her to exclude the possibility that she had mild symptoms from an extreme CTD disease. Defining a precise gene mutation (or mutations) allows preimplantation genetic diagnosis to select embryos without the pathogenic CTD allele,20 but is obviously ethically and medically complex. Embryos with extreme mutations have only potential for severe CTD, while most individuals with CTD have normal lifespans with good, albeit compromised, qualities of life
Despite the complications of prognostic and prenatal prediction, gene testing has significantly improved our understanding of EDS and other CTDs. In 100+ CTD patients who have had WES, the author has found over 20 gene changes that were not previously associated with tissue laxity/fragility or dysautonomia, ranging from those causing acute intermittent porphyria to dopamine-beta-hydroxylase that converts norepinephrine to dopamine. At present, these gene changes do not influence the joint protection or nutritional therapies guided by the clinical diagnosis of CTD-dysautonomia. Yet, a future of preconception and newborn WES will yield large groups of CTD patients with specific gene mutations, allowing tailored management analogous to immune therapies in cancer that target particular tumor genes. Better interventions should greatly improve the quality of life for patients with CTD.
PREVENTIVE HEALTHCARE AND THERAPY FOR JOINT LAXITY SYNDROMES
Joint pain is one of the most common symptoms experienced by patients with joint laxity syndromes due to their hypermobility causing wear and tear on their joints and predisposing them to arthritis. Analgesics can be used to control joint pain in these patients, even when arthritis is present. Immune inhibitors with potentially severe side effects should not be prescribed to patients with CTD, since their wear and tear arthritis due to disability is different from that in inflammatory arthritis (eg, lupus, rheumatoid arthritis). In addition, common sense measures to protect the joints should be recommended; for example, telling patients to favor joint-friendly activities (eg, swimming) over those that tax the joints (eg, long-distance running, gymnastics).
Although it may be tempting for some patients to minimize their physical activity levels, it is important for them to remain active to prevent joint stiffness and pain and to alleviate some of the many other symptoms that accompany EDS and other CTDs (eg, chronic fatigue syndrome, fibromyalgia, IBS, anxiety/depression). Moderate weightlifting and other muscle-building activities can also be helpful, as they strengthen the muscles around the joints and protect them from injuries (and muscle relaxants should be avoided). When injuries occur, the RICE (Rest, Ice, Compression, Elevation) approach can minimize ongoing joint damage. Early orthopedic evaluation of persisting joint pain to exclude tears, dislocations, and fractures is recommended because of susceptibility to joint injury and slow healing.11-13
Dysautonomia is another common problem among patients with joint laxity syndromes, which can manifest as POTS, excessive fatigue, cardiovascular symptoms (eg, tachycardia, bradycardia, angina), IBS, and other symptoms. Although not validated by controlled trials, several types of therapy are available for dysautonomia, depending on the patient’s presentation. Nutrition strategies include adequate fluid intake (at least 8 glasses daily), abundant salt intake (many patients desire salt because of their orthostatic hypotension), and a daily multivitamin and mineral preparation. In addition, extra vitamin C (500 mg to 2 g daily), vitamin D (>1000 units daily), and B12 (2.5 mg daily) is often recommended. IBS symptoms can be improved by avoiding fluids before meals, eating frequent small meals, using enzyme supplements before each meal, and lowering intake of gluten, dairy, and other foods that are difficult to digest. Biofeedback and cognitive-behavioral therapy approaches can train individuals to compensate for the dizziness and fatigue associated with POTS by interpreting early signals and substituting positive actions and attitudes.
When cardiovascular-related symptoms are present, medication prescribing is best left to the cardiology specialist. However, agents to consider can include beta-blockers or midodrine to counteract sympathetic stimulation, fludrocortisone to aid salt/water repletion of intravascular volume, clonidine for sleep, and assorted medications to control headache, arthralgia, neuralgia, myalgia, and soft tissue and/or visceral pain. Patients who have posterior head and neck pain may benefit from upright head MRI to exclude a Chiari deformation arising from a distensible fourth ventricle and/or cervical spine changes; surgical treatment can be very effective for Chiari deformation.1
CONCLUSION
Many geneticists, physicians, and other healthcare providers continue to view EDS and other CTDs as a group of rare and extreme disorders, only making the diagnosis when very unusual skin or joint findings are observed. Milder forms of these diseases, however, are more common, their associated dysautonomia and chronic pain often misdiagnosed as chronic fatigue syndrome, fibromyalgia, or anxiety. Recognition that these symptoms reflect an underlying, truly medical condition offers validation to patients who are often dismissed by multiple specialists, providing important opportunities for prevention and therapy. When patients present with recurrent joint injuries and/or pain, joint hypermobility should be considered and is easily evaluated.
A major criterion for diagnosing joint hypermobility is a positive Beighton score (≥4 out of 9), and when joint hypermobility occurs in combination with 2 or more of the criteria outlined in the Table, a joint laxity syndrome is likely. Such syndromes are genetic, often with environmental factors as triggers for severe symptoms, and genetic testing using WES is greatly expanding the list of genes that when altered can cause CTD. At present, gene or DNA testing has limited advantages over the clinical CTD diagnosis in terms of guiding interventions or subspecialist referrals. Nevertheless, it can help determine the risk of transmitting the disease to offspring and in the future may lead to more effective treatments. Until then, treatment of patients with EDS and other CTDs focuses on prevention of injuries and managing associated symptoms to improve quality of life. ν
Golder N. Wilson, MD, PhD, is clinical professor of pediatrics at Texas Tech University Health Sciences Center, Lubbock and KinderGenome Genetics, Medical City Hospital in Dallas, TX.
References:
1.Steinman B, Royce PM, Superti-Furga A. The Ehlers-Danlos syndrome. In: B Steinman, PM Royce, eds. Connective Tissue and its Heritable Disorders. New York, NY: Wiley-Liss; 1993:351-407.
2.Kirk JA, Ansell BM, Bywaters EG. The hypermobility syndrome. Musculoskeletal complaints associated with generalized joint hypermobility. Ann Rheum Dis. 1967;26(5):419-425.
3.Remvig L, Jensen DV, Ward RC. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol. 2007;34(4):804-809.
4.Simpson MR. Benign joint hypermobility syndrome: evaluation, diagnosis, and management. J Am Osteopath Assoc. 2006;106(9):531–536.
5.Pyeritz R. A small molecule for a large disease. N Engl J Med. 2008;358(26):2829-2831.
6.Malfait F, De Paepe A. Molecular genetics in classic Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2005;139C(1):17-23.
7.Islam S, Chumak J, Wilson GN. A pregnant woman with joint and skin laxity (Ehlers-Danlos syndrome). Consultant. 2008;48(8).
8.Castori M, Morlino S, Celletti C, et al. Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers-Danlos syndrome, hypermobility type): principles and proposal for a multidisciplinary approach. Am J Med Genet A. 2012;158A(8):2055-2070.
9. Castori M, Morlino S, Celletti C, et al. Re-writing the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers–Danlos syndrome, hypermobility type. Am J Med Genet A. 2013;161A(12):2989-3004.
10. Gazit Y, Nahir AM, Grahame R, Jacob G.Dysautonomia in the joint hypermobility syndrome. Am J Med. 2003;115(1):33-40.
11. Pizzo PA. Lessons in pain relief—a personal postgraduate experience. N Engl J Med. 2013;369(12):1092-1093.
12. Bathen T, Hangmann AB, Hoff M, et al. Multidisciplinary treatment of disability in Ehlers-Danlos syndrome hypermobility type/hypermobility syndrome: a pilot study using a combination of physical and cognitive- behavioral therapy on 12 women. Am J Med Genet A. 2013;161A(12):3005-3011.
13. Murray B, Yashar BM, Uhlmann WR, et al. Ehlers–Danlos syndrome, hypermobility type: a characterization of the patients’ lived experience. Am J Med Genet A. 2013;161A(12):2981-2988.
14. Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369(16):1502-1511.
15. Ehlers-Danlos Syndrome National Foundation. www.ednf.org/. Accessed January, 2015.
16. Wilson GN, Cooley WC: Preventive Health Care for Children with Genetic Condition: Providing a Medical Home. 2nd ed. Cambridge, MA. Cambridge University Press, 2006.
17. Wilson GN. Exome sequencing analysis of connective tissue dysplasia: death and rebirth of clinical genetics? Am J Med Genet A. 2014;164(5):1209-1212.
18. Online Mendelian Inheritance in Man (OMIM). www.omim.org. Accessed October 28, 2014.
19. GeneTests© (www.genetests.org), accessed January, 2015.
20. Wilson GN. Presymptomatic and preimplantation genetic diagnosis: neurology, NextGenetics, and the next generation. JAMA Neurol. 2014;71(4):403-404.